




Discover more from Holodoxa
The Tumor Suppressor to Rule Them All
A story about a gene you should know about, TP53, and its associated cancer syndrome.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
In the mid-1970s, cancer biology began to coalesce as a fruitful clinical and basic science. One of the early influential findings concerned a discovery made in 1976 by a team of scientists, Dominique Stehelin, Harold Varmus, J. Michael Bishop, and Peter Vogt.1 They identified a gene from a retrovirus known to cause tumors in chickens. To their surprise, this gene (Src) also was present in the genomes of chickens. Something about the infective delivery of the Src gene was inducing the growth of tumors.
This was the first discovery of an oncogene, an important concept in cancer biology that would blossom as cancer increasingly became understood as the loss of a normal genetic program in cells. At the time, though, this discovery naturally directed cancer researcher attention to viral pathogens. A viral theory of cancer existed before this discovery and thus many picked up the ball and ran with it. Today, we know that viral infections account for a small portion (roughly 4%) of annual cancer incidence.2 Nonetheless, this research was critical to the foundations of modern cancer biology. In fact, it contributed to the crucial discovery of the most important tumor suppressor gene in the genome, TP53.

Oncoviruses Expose the Guardian of the Genome
TP53 is often referred to as the “Guardian of the Genome.” This grandiloquent moniker is well earned. It is frequently mutated in cancer. The generously ballparked figure is usually around 50% with some variance between different cancer types. Plus, the presence of mutation in TP53 is often associated with a poor prognosis. This is a big deal in terms of the somatic landscape of cancer. There isn’t any other gene anywhere close to as frequently altered (see the table below). This places TP53 at central command of the normal cellular program. Before we explore why this is, let’s return to how pioneering cancer biologists discovered TP53.
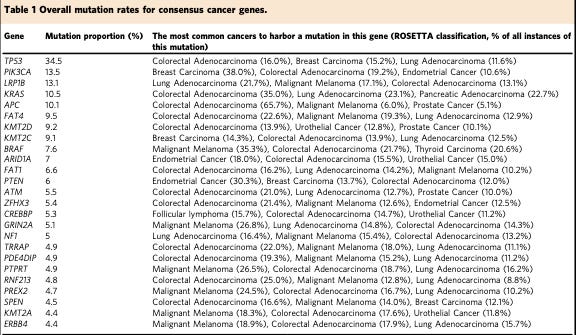
Back in the late 70s, a number of cancer biologists were interested in an oncovirus called SV40. While studying the sera of rabbits with SV40-associated tumors used to identify SV40 antigens from the lysate (the broken open and isolated contents) of SV40-infected mouse cells, these scientists stumbled across a protein with the apparent molecular mass of 53 kiloDaltons (kDa).3 They gave the protein an uninspiring descriptive name p53 or protein of 53 kDa.
This p53 was a bit of a mystery, but the tinkering cancer biologists continued to stumble across it in other virally transformed cells. They found that it was elevated in these contexts and that the viral proteins often bound to it. Its repeated appearance in experiments inspired scientists to map its genomic region and clone the gene to learn more about it. These efforts came to fruition in the mid-1980s.
TP53 was mapped using a hybridization approach: human-rodent cell hybridization, in situ chromosomal hybridization, and direct DNA probes derived from mice.4 Hybridization studies take advantage of the natural complementarity of DNA.5 In these approaches, there is a target sequence and a bait sequence. The bait sequence is designed to match the target and is primed with some modification that enables visualization of where the bait has taken. The scientists purposefully chose baits derived from the RNA of SV40-infected cells. They already knew TP53 was highly expressed in these context and thus smartly guessed the RNA would be a useful source of a bait. The RNA of a gene is a natural complement of the DNA gene it is transcribed from and can be converted back into DNA using a special enzyme and modified nucleotides. The modified nucleotides at the time were radioactive because the radioactivity enabled detection of where the probe landed.6
What is p53? Anything but a Simple Protein
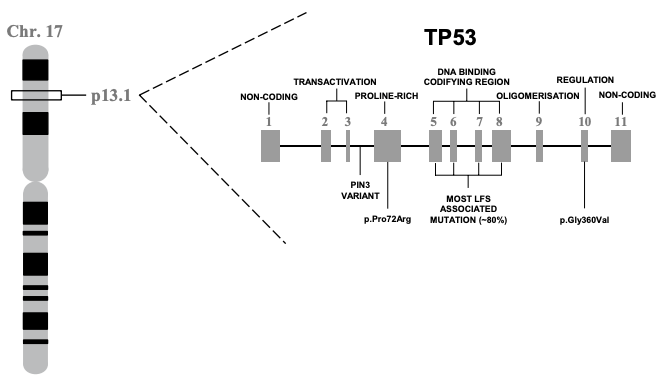
In 1985/6, TP53 was finally located on the distal region of the short arm of chromosome 17 (17p13), spanning a 20 kilobase (kb) region. Concurrently, the gene was cloned for molecular study. This work showed that TP53 is comprised of 11 exons of which the first was entirely non-protein-coding and situated 8 – 10 kb away from the remaining exons. Together these 11 exons produce a protein 393-amino acids in length. There are several functional domains to p53 but the transactivation domain (residues 1-64) and the DNA-binding domain (residues 94 – 292) are probably the most critical for understanding its molecular function. Cross-species analysis of the gene sequence demonstrated that TP53 is unsurprisingly a highly conserved gene, especially the amino acid residues 13 – 23, 117 – 142, 171 – 181, 234 – 250 and 270 – 286. Additional evolutionary analyses suggest the gene evolved some 600-800 million years ago. The cross-species similarity and the deep history of the gene make sense given how important p53 is in terms of keeping cell behavior in check.

After more than forty years of intensive research and because of millions of years of evolution, numerous functions have been described for or ascribed to p53. We now recognize it as a critical node in many cellular processes related to the response to cellular stress: DNA damage, hypoxia, oncogene addiction, and aging. The cell biology textbook lesson on p53 is typically that it is the cell’s rapid response mediator of the DNA damage response, directing repair or pushing the cell toward self-destruction (apoptosis) if the damage is overwhelming. The second part of this textbook lesson covers how this response is calibrated by a ubiquitin ligase protein called MDM2. In a normal cellular states MDM2 keeps levels of p53 low, but when stress strikes, MDM2’s regulation is released and p53 drives a feedforward response to the stress.7

But What About the Germline? TP53 and Li-Fraumeni Syndrome
As we’ve seen, TP53 is enormously important to keeping cell behavior copacetic, especially when things go crazy. This is important to the story of how cells lose control as we get older. Losing p53 in the battle with father time is almost assuredly a cancer diagnosis in the making. But this is a series on hereditary cancer after-all, and there are people who have the misfortune of inheriting mutations in the genome’s guardian.
Those born with mutations in TP53 can present heterogeneously, but they usually end up with a clinical diagnosis of Li-Fraumeni syndrome (LFS). LFS shows an autosomal dominant pattern of inheritance, indicating that only one mutation is necessary to cause disease and carriers have a 50% risk of transmitting the mutation to offspring. Inherited or de novo mutations in TP53 represent about 70% of classic LFS cases, though no other genetic relationships have been validated. CHEK2 mutations have been observed in LFS but in less than a handful of reports. Classic LFS is clinically defined as a sarcoma before 45, a first-degree relative with cancer before 45, and an additional relative (first- or second-degree) with any cancer before 45 or sarcoma at any age. However, clinical criteria for diagnosis can vary, having relaxed a bit overtime. An expanded clinical entity related to LFS called Li-Fraumeni-like syndrome (LFL) gets applied to some carriers who come close to but don’t meet the more stringent criteria. Because of this ambiguity, some have proposed that “TP53 Cancer Predisposition Syndrome” supplant LFS and LFL in a similar way to how PTEN Hamartoma Tumor Syndrome came to encompass cancer syndromes like Cowden syndrome when PTEN mutations were present. As research progresses, the critical clinical information is increasingly the molecular diagnosis, the finding of a germline TP53 mutation.
TP53 was first reported in connection with LFS in 1990 and numerous reports have consistently confirmed this observation. Now, we know that lifetime cancer risk for individuals with these mutation is greater than or equal to 70% for men and 90% for women. These cancers tend to present quite early in life as well, at least half before 30. These estimates of disease penetrance and severity may be overestimated because we don’t necessarily have a great understanding of how common pathogenic variation in TP53 is. In other words, these figures may be somewhat skewed by ascertainment bias; those most affected show up for assessment. The spectrum of cancer risk is distributed across many tissues but five major types explain the lion’s share of LFS-associated cancer: adrenocortical carcinomas, breast carcinomas, central nervous system tumors, osteosarcomas, and soft-tissue sarcomas. Due to this very high cancer risk and the fact that many LFS patients exhibit multiple primary tumors, certain preventative measures are strongly recommended along with an intensive surveillance schedule.
A number of different TP53 mutations have been described in LFS patients, including missense, small duplications, small deletions, frameshift, and nonsense mutations.8 Although the prevalence of TP53 mutations in the germline is not definitively pinned down, the estimates (1:3555 to 1:5476) suggest that it is modestly prevalent. Much more common than mutations in PTEN (1:200000) but somewhat less common than BRCA1 (1:400). Moreover, de novo inheritance is especially notable, possibly accounting for 20% of LFS cases. There aren’t currently any great explanation for this high rate of spontaneous new mutations in the gene. However, all of these mutations cause disease by destroying the function of the gene (loss-of-function) and sometimes even working to interfere with the function of the other healthy allele (dominant negativity). Having less p53 around in the cell means that the cell cannot respond to stressors like DNA damage properly. This situation in cells is only tenable for so long before cancer processes take off.
Genotype-Phenotype Research Plodding Along
For my much appreciated loyal readers following the Cancer Genomes series, it should be apparent that heterogeneity (a diversity of symptoms from seemingly the same cause) is a clinical challenge and a urgent research question when it comes to cancer syndromes. Why does one genotype associate with such a broad range of phenotypes, including different cancers? I previously tried to examine some possible explanations for the organ-specific patterns of disease associated with BRCA mutations, but much remains unknown. Unfortunately, the same is true for TP53 and LFS.
To solve this problem, scientists have in part turned again to multiplex assays of variant effect (MAVE). Four MAVEs have been conducted on TP53 but have been published in two different papers. One was published in Nature Genetics and the other in Science, very prestigious journals - a signal of the expected impact of the research. The upshot of these studies is that they likely improve the interpretation of the pathogenicity (or disease-causing-ness) of any given change to the gene. In fact, a follow-up study was able to integrate the datasets from these four MAVEs and generate evidence to help reclassify TP53 variants. Hopefully, this work helps catalyze future research that definitively sorts out questions about genotype-phenotype relationships. This will enable advanced precision care to be delivered to those with LFS.
Varmus and Bishop would go on to win a Nobel prize in physiology and medicine for this and related work in 1989.
This figure comes from a recent meta-analysis published in JAMA Oncology. The authors found ~4% of cancers diagnosed in adults in 2017 were attributable to viral infections. The responsible oncogenic viruses in order of their contribution were HPV, HCV, EBV, HBV, MCPyV, KSHV, & HTLV-1. This number isn’t insignificant but for context it is dwarfed by cancer risk associated with inherited cancer syndrome which may represent about 10% of cancer cases. Furthermore, it is likely that the share of cancers subsequent to viral infections will decline due to improved public health measures and other prevention strategies (like vaccines).
A Dalton is a non-SI unit of mass used to express molecular mass of large molecules, usually proteins. 1 Da is equivalent to 1 gram per mole. Thus, p53 is a protein of the mass 53,000 grams per mole.
In previous Cancer Genomes posts, gene mapping was typically carried out via genetic linkage analysis. In this case, the gene product was already known and some molecular understanding already existed about p53. This enabled a simpler and more direct approach. Additionally, the linkage work on the genetic syndrome associated with germline TP53 mutations was not mature until the early 2000s.
DNA is double-stranded molecule, where one strand has a given set of nucleotides (called bases) in a particular direction and the other strand has different yet complementary set of nucleotides running in the other direction. Probes can be designed as a complement of a given sequence.
Today, we probably wouldn’t use radiation but rather chemical modifications that emits particular wavelengths of light when excited.
It is considered a positive feedback loop because p53 drives its own expression. It helps make more of itself.
There are databases that describe TP53 variants of interest in Li-Fraumeni Syndrome, including: LOVD, The TP53 Website, the IARC TP53 database, the Database of germline p53 mutations, and the UMD TP53 website.
Subscribe to Holodoxa
A study of the total human condition. I examine ideas from science, literature, technology, politics, and more. Most posts will consist of book reviews or short essays.








