




Discover more from Holodoxa
Praetorian PTEN: Protecting Cells from Cancer
Holodoxa's Cancer Genome series continues. Part 2 on PTEN, exploring its function in cells and why it is important to stopping cancer.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
Sometimes when I think about PTEN, I recall a scene from the television series A Game of Thrones. In my mind’s eye, I see Jon Snow grimly standing alone before an oncoming rush of enemy soldiers on horseback just as PTEN is the bulwark against the proto-oncogenes poised to wreck havoc in cells and drive cancer growth.

But just like this gif, when PTEN is working, it inspires the cell’s other actors to array themselves against the forces of uncontrolled growth and proliferation and beat them back. This is why cancer scientists will often refer to PTEN as a “master regulator” of of cell growth and proliferation. It is also why when the function of PTEN is lost because of mutation, whether inherited or sporadic, cancer is often the consequence.
But enough for the imperfect pop culture analogy, let’s jump into the biology!
Structure and Function
As mentioned in my first post on PTEN, it is a gene that encodes a “dual-specificity phosphatase.” This is opaque biochemical jargon meant as a pithy description of the primary function of PTEN. The whole story involves a lot more explanation, but the arc of how we go from PTEN the gene to PTEN the protein is a pathway referred to as the “central dogma” of biology. The DNA sequence of a gene is copied down or “transcribed” by cellular machinery1 into a messenger molecule comprised of RNA. Then, the messenger molecule travels to another part of the cell where different cellular machinery2 “translates” the RNA into protein. The protein then snaps from a long chain of amino acids into a three-dimensional protein that can carry out various actions. For proteins that float around in the cellular goo like PTEN, they usually perform some sort of biochemical reaction. In the case of PTEN, this is removing a chemical entity called a phosphate group from specific fats and proteins. The fact that PTEN can do this to both fats and proteins is why its phosphatase function is referred to as “dual-specificity.” Later, we’ll return to why the biochemical activity of PTEN is actually important to cellular states.
Now that we’re acquainted with what PTEN does, let’s examine its structure. A tenet of biology is that structure reveals function. This is essentially the reciprocal of the architectural adage of “form follows function.” Subsequently, there’s a long tradition of studying the ways that biomolecules are physically and chemically organized. At least four Nobel prizes that mark pivotal moments in the history of biomolecular science have been awarded on this basis: DNA (James Watson & Francis Crick), myoglobin & hemoglobin (John Kendrew & Max Perutz), cellular substructures (Albert Claude, Christian de Duve, & George E. Palade), and the CRISPR guide RNA (Emmanuelle Charpentier & Jennifer Doudna).
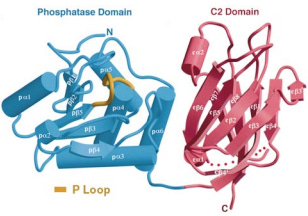
At the genome-level, PTEN stretches across 105 thousand DNA base pairs on the long arm of chromosome ten. It has 9 protein-coding segments called exons that are punctuated by 8 non-protein-coding segments called introns. When PTEN is transcribed into RNA, there is cellular machinery3 that removes introns and stitches together the exons. Once this is complete, the coding sequence of PTEN is now 1212 bases of RNA long and can be read unidirectionally4 by the cellular machine that translates it into protein, the ribosome. After translation, PTEN is now a polypeptide (aka protein) of 403 amino acids. There are four functional regions or “domains” of PTEN. At the very beginning, there is an unstructured, meaning non-folded region, that is important to where PTEN get directed to go in the cell. The second and most glamorous domain of PTEN (shown in blue below) is the protein tyrosine phosphatase domain (abbreviated as PTPase)5. This is followed by a second structural region called the C2 domain (shown in yellow), which helps anchor PTEN on the inner leaflet of a cell’s lipid bilayer and facilitates the removal of the phosphate at the third inositol ring position from a lipid called phosphatidylinositol-3,4,5-triphosphate or PIP3. And the final domain is an long unstructured tail that is subject to modification by other proteins, especially kinases, and also helps localize PTEN to certain places in the cell. Kinases are essentially the cellular opposites of phosphatases. They add phosphate groups to certain amino acids on proteins, which are represented as green P’s in the figure below.

Shortly after PTEN was discovered as mutated in sporadic cancers and as a risk gene for Cowden syndrome, an analysis of the three-dimensional structure was published in the prestigious journal Cell by a group led by Pier Pandolfi and Nikola Pavletich. This proved a difficult project for two related reasons. One, there are unstructured regions of PTEN, which prevent PTEN from crystalizing or “freezing” in solution. Two, at the time, the only method to comprehensively map the three-dimensional or “crystal” structure of PTEN at high resolution was X-ray crystallography, which as the name suggests required crystals. In fact, the only particularly finicky part of an crystallography analysis is making the crystals.
To get around this issue, the authors of the crystal study removed the unstructured regions and created an entirely new PTEN protein. Thus, knowledge of the 3D structure of PTEN is still primarily informed by this 1999 study and only provides coverage of the two main functional domains of PTEN, PTPase and C2, in their crystallized configurations. Unfortunately, PTEN is also poorly suited to structural study by modern alternatives to X-ray crystallography like cryo-electron microscopy for other reasons (mainly its size). Subsequently, scientists interested in learning more about the functional consequences of genetic variation on PTEN structure and function have turned to alternative methods like biophysical simulation and deep mutational scanning. Much remains to be learned, including whether PTEN can be targeted with drugs.
How Does a Little Phosphatase Hold Cancer Back?
Despite the limitations on structural analysis, research scientists have still learned a great deal about PTEN from other types of empirical work. The big contributions have drilled down on how the loss of PTEN function impacts cellular behavior. These are the studies that have placed PTEN in the pantheon of important tumor suppressor genes along with the guardian of the genome TP53 and the textbook case RB1 (both important genes in hereditary cancer that we’ll touch on later).
The lion’s share of PTEN’s anti-growth effects is attributed to its lipid phosphatase activity. Again, PTEN removes a specific phosphate group from a fat molecule called PIP3. It just so happens to be that PIP3 is a crucial lipid messenger, a signal in the cellular noise. The amount and location of PIP3 in a cell’s membrane influences growth, proliferation, and migration (stuff cancers like to do). It triggers a whole cascade of proteins that coordinate important pro-growth actions. This signaling cascade is called the PI3K/AKT/mTOR pathway. It is one of the primary mediators of growth and proliferation. In fact, mTOR is considered the central mediator of metabolic pathways that build the cell (anabolism). In healthy growth conditions, cells depend on the cellular actions of PI3K/AKT/mTOR, all kinases, to directed growth and division. But, of course, there can be too much of a good thing. PTEN enforces moderation, retrains the unruly horde of pro-growth kinases.

In general, kinases and phophatases are, respectively, the yin and yang of cellular growth. This make interpretation of a given perturbation in any single node in the pathway fairly straightforward. There is a “GO” side (PI3K/AKT/mTOR) and a “STOP” side (PTEN/TSC1/2).6 If the “STOP” side fails, the “GO” side drives cancer. So the hard part isn’t really the why now that the pathway is well described, but the how regulation of the pathway fails. How much dysfunction in PTEN is required to tip the cell into tumorigenesis? And is PTEN dysfunction alone sufficient to drive tumorigenesis? The genetic evidence strongly indicated a yes on the first question, and we have a reasonably good answer for question too. However, it took years of careful experimentation and an expansion of the basic paradigm of how cancer starts.
A Continuum Model for Tumor Suppression
The “how” question is perpetually a lodestar for biomedical scientists. The ability to effectively and reliably intervene on some disease biology often depends on a deep mechanistic understanding on that phenomenon. Fortunately, the field of cancer biology and the study cancer syndromes has made strides on this front. Our understanding of how cancer starts, especially in individuals who inherit cancer risk mutations, has advanced respectably. It is at least a the point where every undergraduate cancer biology textbook has a story to tell about it. That story of cancer initiation is what’s known as the Two-Hit Hypothesis.

The Two-Hit Hypothesis presents a paradigm based on the fact everyone inherits two copies (alleles) of a gene from their mother and father. But one inherited mutation in a tumor suppressor gene isn’t enough to initiate cancer. Patients with germline PTEN mutations aren’t born with congenital cancers. It takes years for cancer to appear and for many familial cancers this can take well into adulthood. In order to explain this delay, Alfred Knudson argued that there must be some second event in the remaining healthy copy of the tumor suppressor gene (aka two hits) that then drives a healthy cell to become cancerous. In this model, inherited cancer risk isn’t mediated by the inherited mutation itself but by the lack of two healthy copies to absorb random mutational events. The probability of cancer is turned way up by the initial lesion; the patient goes from having two brake pedals on cancer to just one and one isn’t enough over time.
However, there was a problem. When it came to finding second-hits in the cancer of patients with PTEN mutations, none were turning up (this has recently changed, but it is still generally true). The Two-Hit model wasn’t useful for explaining tumorigenesis in PHTS patients. Instead, it took careful experimentation where Pten expression could be precisely turned down in a mouse model to see what was the minimum amount of Pten necessary to keep tumorigeneis at bay. This work was used to support an alternative model of tumor suppression: The Continuum Model.
The Continuum Model was first described in a letter to the prestigious journal Nature in 2011, marking the 40th anniversary of the first evidence for the Two-Hit Hypothesis. Authored by Dr. Pandolfi with Drs. Alice Berger and Alfred Knudson, the Continuum Model argued that the classical Two-Hit Hypothesis, which states that cancer develops when both copies of a tumor suppressor gene are mutated or lost, is too narrow and does not account for the full spectrum of tumor suppressor gene biology. Instead, they posited that tumor suppressor genes experience a continuum of functions that depend on their dosage, expression, activity, and interactions with other genes and pathways. They also discuss some exceptions to the Two-Hit Hypothesis, such as cases where partial loss of a tumor suppressor gene is more harmful than complete loss, or where tumor suppressor genes have different roles in different tissues or stages of development. The study of how PTEN mutations drive cancer helped pioneer the Continuum Model and now is the model’s canonical example.

Next in Cancer Genomes
This concludes the second installment in the hereditary cancer series. There will be one final post on PTEN to dig into some of the latest genetic research, especially attempts to learn more functional information about the consequence of variation in PTEN and some other interesting PTEN biology.
I am referring to the proteins involved in transcription, namely RNA polymerase, which is the enzyme responsible for converting the gene sequence into an RNA molecule.
Here, I am referring to ribosomes, which are a complexes of RNA and protein that coordinate the translation of the RNA message into a protein.
This refers to the spliceosome, another protein and RNA complex. Splicing actually occurs co-transcriptionally, meaning while the DNA is being actively converted into an RNA message the spliceosome is furiously working to remove intron sequences and join together exon sequences. Splicing is an important mechanism for generating a diversity of protein forms from a single gene. There is a rough correlation between organismal complexity and splicing complexity.
DNA synthesis, RNA transcription, and protein translation all occur in the 5’ (read “five prime”) to 3’ (read “three prime”) direction. 5’ and 3’ refer to the location of hydroxyl groups (-OH) on the sugar backbone of DNA/RNA.
Even though PTEN has dual specificity in its choice of substrate, it is classed as a protein tyrosine phosphatase, meaning it remove phosphates from tyrosine amino acid residues. It is classed this way based on conservation analysis of its sequence, which attempt to trace the evolutionary origins of a given gene sequence. There are six subfamilies of dual-specificity phosphatase of which PTEN itself defines a class that includes four other genes: TNS1, TNS2, TPTE, and TPTE2.
This harks back to the gas vs brake metaphor for oncogenes and tumor suppressors from part 1 on PTEN and Cowden/PHTS.
Subscribe to Holodoxa
A study of the total human condition. I examine ideas from science, literature, technology, politics, and more. Most posts will consist of book reviews or short essays.






