




Discover more from Holodoxa
A Dormant Army Within
How we learned to unleash T cells on cancer.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
The human body is equipped with a suite of cells that protect it from external insults, the immune system. This system naturally partitions immunity into roughly two functional types, innate and adaptive. Innate immunity is provided for by cells like macrophages that can chew up debris and obvious pathogens. Adaptive immunity is provisioned by T and B cells which coordinate cellular and humoral immunity, respectively. The immune system is our powerful ally but can sometimes falter. It can overreact (hypersensitivies/allergies), attack us (autoimmunity), or fail to respond when needed (immunosuppression). One of the high impact points of failure points has been in cancer.
Scientists and physicians have long wondered how cancer evades notice by the immune system and whether the immune system can be directed to kill cancer. In the last decade, this tantalizing theoretical possibility became a reality. We now have an armamentarium of therapeutics that activate latent immunity against cancer.1 In many cases, there are miraculous responses to such approaches. This was made possible by brilliant scientists who elucidated the complex biology underlying cancer’s immune evasion and cellular immunity broadly. In 2018, the two scientists, James Allison and Tasuku Honjo, were credited with the “discovery of cancer therapy by inhibition of negative immune regulation” and awarded the Nobel prize in Physiology or Medicine. Let’s get acquainted with them and their science.
Co-Stimulation & Co-Inhibition - The Complexity of T Cell Regulation
In 1996, a concise research paper from the Berkeley laboratory of immunologist James Allison was published in Science magazine. At the time, it was already understood that T cell2 activation required the binding of the T cell receptor (TCR) to an antigen3 during antigen presentation. However, it was also known that this understanding of T cell activation was incomplete. There was a missing puzzle piece. The findings from Allison’s lab helped complete the immunological puzzle, expanding our understanding of T cell regulation. Allison found that a protein called cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibits T cell activation. The study built on existing ideas about the phenomenon of T cell co-stimulation, the idea that stimulation of receptors (often called B7 type receptors) in conjunction with the TCR stimulation are necessary to achieve full T cell activation.4 Allison’s research showed that CTLA-4 appeared to block normal co-stimulation and that normal co-stimulation of T cells in a cancer microenvironment could be restored by blocking CTLA-4.
This finding turned out to be momentous. It was the fruit from Allison’s persistent interest in the immune system and cancer. His research was animated by the nagging question, “how do cancer cells evade the immune system, especially as they progress from a primary tumor to advanced metastatic disease?” The CTLA-4 finding was a critical insight.5 It was a proof-of-concept that cancer could be treated via the immune system. This research fundamentally enabled medicine to tweak the immune system to fight cancer.
For those unfamiliar with immunology and cancer biology, some of what’s above is almost certainly gobbledygook. I’ll try and remedy this. Fortunately, we can quote the 1996 paper to aid this effort. It is fairly accessible, which surprisingly is often a feature of many great scientific papers. Thus, we can lift from Allison’s conclusions directly. He sums up the key insight with clarity, brevity, and minimal jargon: “[E]ven though most tissue-derived tumors may present antigen in the context of MHC molecules, they may fail to elicit effective immunity because of a lack of co-stimulatory ability.” In other words, Allison is saying that the presentation of tumor antigens to the immune system6 is not enough by itself to provoke the immune system to attack and kill the tumor. There needs to be additional signaling, co-stimulation. Finding the CTLA-4 was actually an additional layer of complexity beyond co-stimulation though. CTLA-4 is actually an inhibitor of co-stimulation (co-inhibition) and thus an inhibitor of T cell activation. This meant that blockade of CTLA-4 is an attractive option to re-enable the normal co-stimulation process and enable T cells to attack tumors.

Immune Checkpoints - The Programmed Death Story
Around the same time but across the globe from Allison’s laboratory, Tasuku Honjo had been actively investigating an immune checkpoint protein known as programmed death-1 (PD-1). PD-1 was discovered in populations of dying T cells. The expression of PD-1 increased in T cells undergoing apoptosis (programmed cell death), which inspired its name. In a 1992 paper, Honjo’s lab reported that activated T cells exhibit elevated levels of PD-1 on their surfaces. Biochemical studies revealed PD-1 as a transmembrane protein with a genetic fingerprint placing it within the immunoglobulin family. Further research demonstrated enhanced PD-1 expression in activated B cells. Plus, structural analyses highlighted similarities between PD-1 and CTLA-4; both are transmembrane proteins associated with the immunoglobulin family. These were important hints about PD-1’s function.
After sustained follow-up on PD-1, Honjo team finally got what they were looking for - confirmation of another immune checkpoint in T cell regulation. The final bit of their groundbreaking discovery is illustrated in their 2000 paper published in the Journal of Experimental Medicine. Honjo’s team revealed that, like CTLA-4, PD-1 binds to members of the B7 gene family on antigen-presenting cells (APCs). Honjo termed the B7-related binding partners of PD-1 as the ligand of PD-1 (PD-L1). Clever experiments in the same paper demonstrated that PD-L1 binding to PD-1 dampens T cell activation, confirming the PD-1/PD-L1 pathway as an immune checkpoint. Like CTLA4, both PD-L1 and PD-1 have turned out to be bonafide druggable targets.

The drugs that hit targets like CTLA-4, PD-1, and PD-L1 are often called checkpoint inhibitor and are usually monoclonal antibodies. There are numerous agents approved by governnmental agencies like the FDA for the treatment of different cancer types. Responses to these agents can be variable but long-term remissions generally occur in about a fifth of those treated (we’ll turn to the reasons that responses vary next). Numerous clinical trials have shown dramatic effects in cancer patients with new positive results rolling in on a regular basis. Currently, Keytruda (pembrolizumab), an anti-PD-1 antibody by Merck, is the top cancer drug blockbuster drug doing billions in revenue yearly. President Jimmy Carter remains alive and cancer-free today at 99, despite former brain metastases from advanced melanoma, because of drugs like these.

The Landscape of Immune Checkpoint Inhibition
As of the mid-point of 2024, there are over 10 agent approved by the FDA that fit neatly into the immune checkpoint inhibitor (ICI) box. I’ve extracted some tables and figures from various review papers on immunotherapy to provide a recent though not perfectly up-to-date picture of the treatment landscape. As can be surmised quickly from the tables and figures below, these drugs mostly target PD-1 or PD-L1. These targets have definitely shown the most benefit.
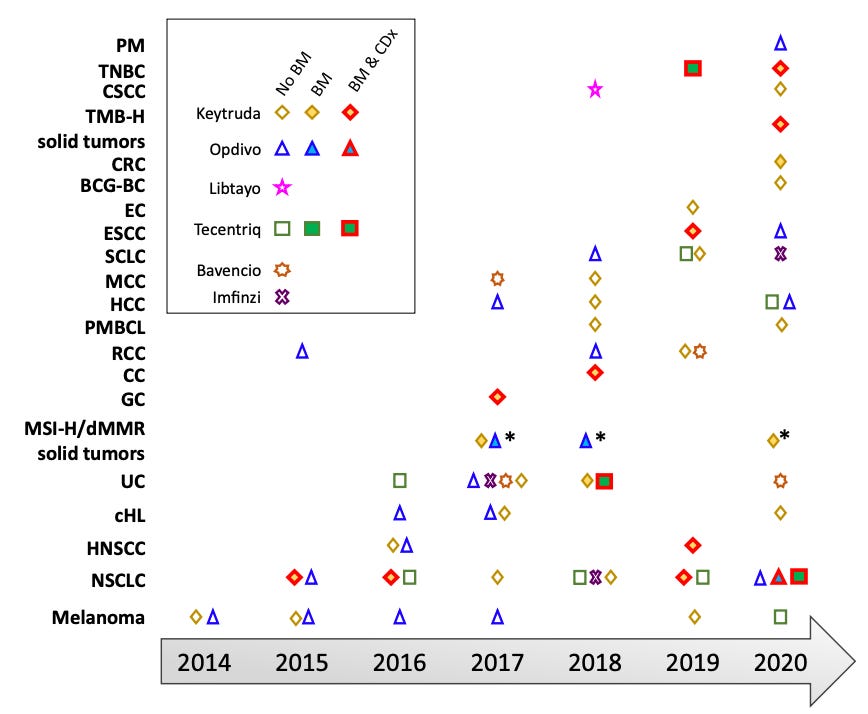

But there’s more. These ICIs can be used to treat many different types of cancer. Non-small cell lung cancer (NSCLC), renal cell carcinoma, and melanoma are some cancer types that see frequent use of ICIs. We can also reasonably expect ICI utilization to continue to increase but that it will be increasingly difficult for oncologist to know what the optimal ICI is for their patient and/or whether to use an ICI for a given patient.
The reasons for this complexity arise from the increasing number of indications (the agency approved use case for a drug) where ICIs are options, the different constraints these indications put on ICI use, and how well these align with day-to-day clinical management of individual patients. First, ICI use is not just available for the treatment of different cancers but also in different settings or lines of treatment. In some cases, ICIs may be an option prior to surgery to de-bulk the tumor (neoadjuvant), after surgical resection (adjuvant), as the first line option in advanced disease, or after one or more treatment options have been exhausted. There are also the complexity of treatment sequencing. For instance, it is often undesirable to give immunotherapy prior to molecularly targeted therapies due to the toxicities that can arise. This is really just scratching the surface of the complexity involved. We have yet to get to the variable biomarker criteria embedded in the indications.
As hinted above, the responses in patients vary substantially. This variation occurs both within and across disease states. We have some idea of the factors that influence responses but much remains unknown. The latest is to use biomarkers of genomic instability: tumor mutational burden, microsatellite status/mismatch repair status, and homologous recombination competence. However, there is still variation in responses even in subset where responses are known to be better (i.e. in those with more genomically unstable tumors). The use of molecular testing relying on next-generation sequencing (NGS) platform will be critical to solving the responder problem in ICIs.
Immunotherapy has unlocked one of the holy grails of cancer treatment - (re)activating the immune system. Immunotherapy may offer promises beyond cancer, including in autoimmune disease. Additionally, we are very much still in the first generation of immunotherapy agent. There will be innovation both within the ICI class and beyond it. Ideally, we can find a solution with near universal efficacy or at least resolves issues with predicting treatment response. Nonetheless, we should all be excited about and attuned to what immunotherapy is and coming developments.

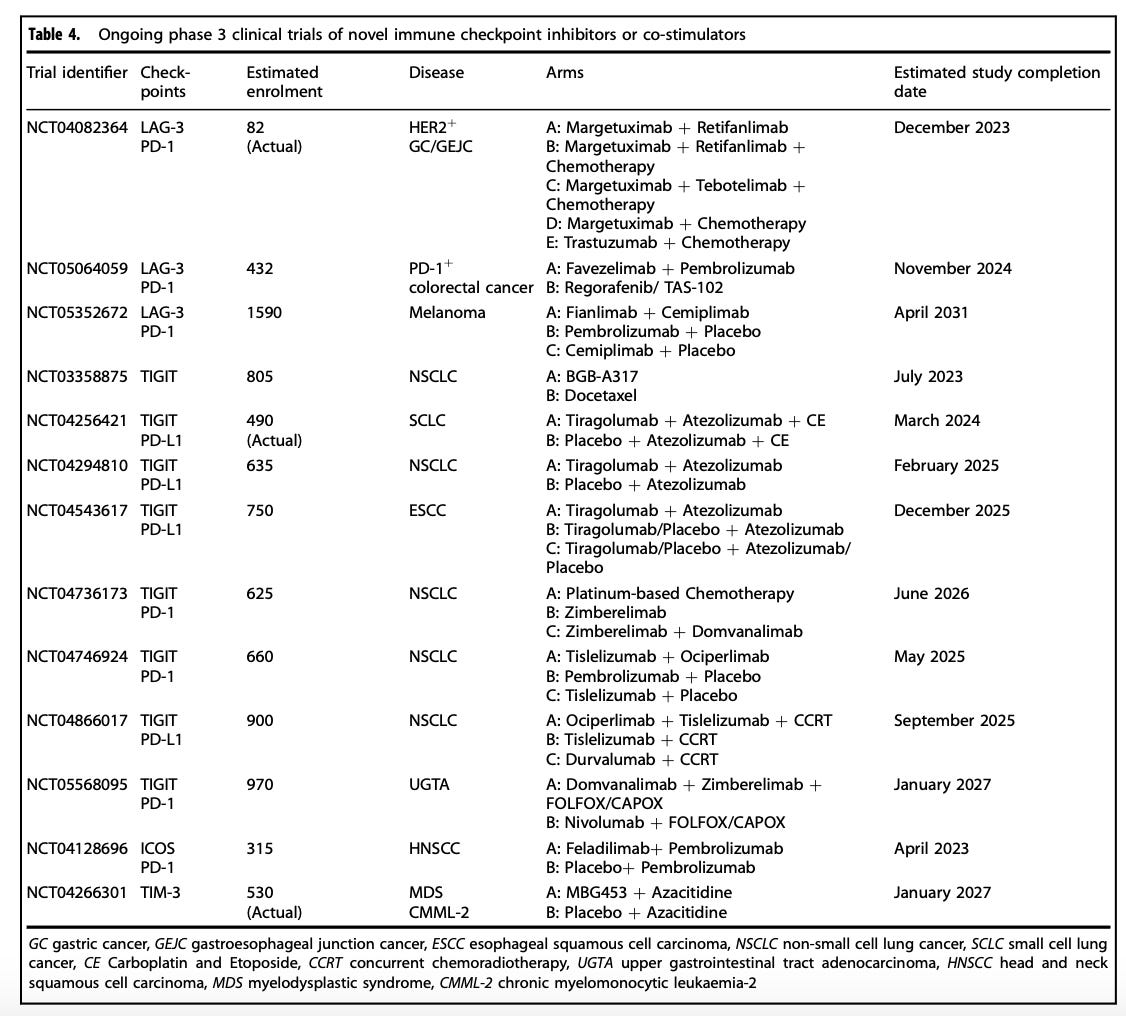
There are even quite crowded immunotherapeutic spaces, depending on the class of immunotherapy we are talking about. In fact, there are now ten anti-PD-(L)1 antibody therapies with FDA market approvals.
An important effector cell of the immune system.
Any substance that provokes a response from the body’s immune system.
As with almost everything in scientific research, this wasn’t the absolute first discovery concerning T cell co-stimulation and there is a lot more to the biology. There was already some previous research that had identified key mechanisms enhancing T cell responses, including co-stimulatory signals mediated by CD28 on T cells interacting with ligands B7-1 (CD80) and B7-2 (CD86) on antigen-presenting cells. Nonetheless, Allison’s finding was pivotal.
What did the 1996 actually show? The Allison lab found that like CD28, the co-stimulatory molecule, CTLA-4 on T cells binds to B7-1, the co-stimulatory receptor, on antigen-presenting cells (the tumor in this case), but with higher affinity. By displacing CD28 from B7-1, CTLA-4 acts as a brake, inhibiting T cell stimulation. CTLA-4 exists because without a stop signal, the immune response could attack healthy cells, leading to autoimmunity and tissue damage. Based on this hypothesized biology, Allison’s team injected antibodies against CTLA-4 in mice with cancer to see if blockade of CTLA-4-B7-1 binding would re-enable T cell activation. And indeed, removing the brake provided by CTLA-4 binding reawakened quiescent T cells into potent tumor killers. Blocking CTLA-4 with antibodies led to increased survival and reduced tumor size in treated animals, while untreated animals showed no response.
This is done with the MHC molecules reference by Allison.
Subscribe to Holodoxa
A study of the total human condition. I examine ideas from science, literature, technology, politics, and more. Most posts will consist of book reviews or short essays.











I hope this is true! I'm on immunotherapy now to prevent cancer recurrence. Pembrolizumab (brand name Keytruda).
I alas failed Keytruda for squamous cell carcinoma of the head and neck, in 2023; right now I'm on an antibody-drug conjugate from Seagen called PDL1V, which, as the name implies, also targets PD-L1: https://jakeseliger.com/2024/05/20/in-which-the-antibody-drug-conjugate-adc-pdl1v-shrinks-the-tumors-in-my-neck-and-buys-me-more-time/. May CT scans showed that it had shrunk tumors; scans from last week, though, appear to show tumors adapting to it as well.