

Why Leukemia Risk is Higher in Hispanic/Latino Children
Why Leukemia Risk is Higher in Hispanic/Latino Children
A case study of an interesting non-coding variant associated with indigenous American ancestry contributing to disparities in acute lymphoblastic leukemia risk.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
Précis
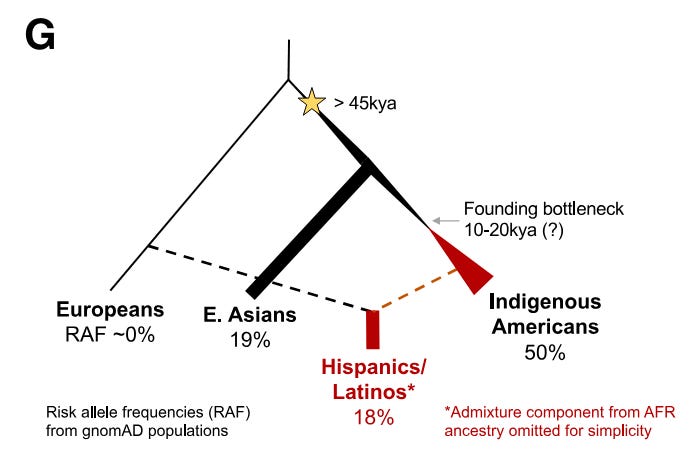
A recent study published this March in the journal Cell Genomics reported a genetic finding that likely explains a meaningful part of the ancestry-associated difference in childhood leukemia. Children from Hispanic/Latino backgrounds, specifically those with admixture from Indigenous American populations, have a higher rate of acute lymphoblastic leukemia (ALL). The authors drew from trans-ancestry genome-wide association study (GWAS) data at a known gene of relevance to ALL, IKZF1, and performed a fine-mapping of variants associated with the GWAS hits. The authors identified a risk variant more than 30 times more common in Hispanic/Latino populations than European populations. They show that positive selection from an unknown cause increased the frequency of this risk variant in Indigenous Americans. They also demonstrate the variant disrupts a region that IKZF1 uses to control its own expression during an important time in the development of B cells. Based on their estimates, this risk variant contributes significantly to the difference in ALL incidence between Hispanics/Latinos and Europeans.
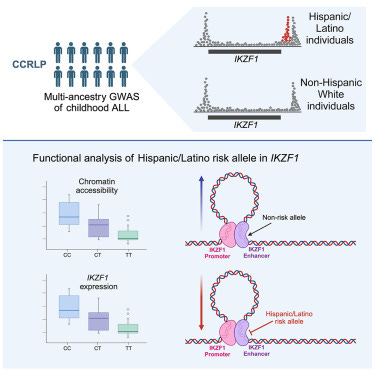
Of all American ethnic groups, self-reported Hispanic/Latino children have the highest risk for acute lymphoblastic leukemia (ALL), the most common childhood cancer. They have a 1.3-fold higher incidence compared to non-Hispanic white children. This difference increases to greater than 2-fold by adolescence and the young adult years.1 This is a bit of an epidemiological mystery. Doctors and scientists have had little in the way to offer as explanation for the disparity. That is until a team of scientists led by Adam J. de Smith, Lara Wahlster, and Soyoung Jeon discovered an intriguing variant in a known risk gene for ALL called IKZF1. Prior research has shown that high-impact rare variants in IKZF1 can cause pediatric ALL, but this is the first connection of a common, like causative IKZF1 allele to ALL risk that also correlates with specific ancestry.2 Broadly, this research underscores the importance of understanding the genetic histories. In this case, the population history of the people who first populated the Americas, those migrating from Northwest Asia between 13-20 kya.3
I don’t want to create a false impression. With any standalone study, this is probably not the entire story. These new findings likely do not explain the entirety of the observed disparity. However, they do suggest that local evolutionary pressures on indigenous Americans thousands of years ago are a critical factor. These data allow us to imagine a more comprehensive explanation, one predominantly rooted in the ancestral history of today’s Hispanic/Latino population. This Cell Genomics study is a clear and thorough illustration of how recent evolutionary selection can cause differential rates of disease today. The actual selection pressure on the IKZF1 variant(s) is not definitively identified, but the authors speculate that infectious agents or other types of immune exposures are likely at play. This is a safe bet given the importance of pathogens in the evolutionary history of all animals.
This type of evolutionary speculation is reasonable as it is orthogonally supported by robust biological evidence. IKZF1 encodes a transcription factor protein that is important to normal B cell development and hematopoiesis. In this same vein, the study provides a clever illustration for how this variant may contribute to ALL risk. The putative casual variant is non-coding and thus doesn’t disrupt IKZF1 function. Rather, it affects its expression at critical moments in the development of B cells. The causal variant does this by disrupting a recursive gene regulatory circuit that is critical to the maturation of B cells. As a carrier of this variant mature, his or her B cells will struggle to differentiate from progenitors cells into precursors and then into mature cells. When B cells are trapped in immature states, they can proliferate and this process can spiral into cancer. Thus, being a carrier raises ALL risk.
I find this work incredibly intriguing. It is always tantalizing to see the potential consequences of evolution at work. Plus, this research nicely focuses on a specific time and place in the history of human populations and identifies a gene variant with a clinically meaningful biological effect. And all of this is downstream of some unknown selection pressure. Examples like these are uncommon.4 Although this is not the most robust demonstration of actual selection, it is an excellent illustration of how differential disease risk across identifiable populations can arise from differences in genetic ancestry. Findings like these are important for a number of reasons. They highlight the importance of genotyping diverse populations of both the past and present.
Sequencing diverse populations seems like an obvious call to me, but there has been criticism of such efforts or at least how the data from such efforts is presented. Some academic geneticists, philosophers of science, and online activists fear these findings can be misused to reify notions of race as a biological phenomenon. However, this study is a deft rejoinder to such concerns in so far as it demonstrates the medical importance of genetic ancestry. I think the value of such knowledge is clear - improving human health and deepening our understanding of our origins, this knowledge far outweighs concerns about exhuming dubious ideas from the ghosts of science’s past.5 Moreover, this is a type of health disparity that is liable to be thoughtlessly chalked up to environmental differences. If we continue to chase answers in the wrong place, we waste precious resources to no avail. We also fail to help those we’re claiming to protect.
How did our sharp-witted scientist know to look at genes for the cause of differential ALL risk? Well apart from this being something geneticists are disposed to know, there were a number of hints that Hispanic enrichment of ALL may be genetic. First, these cases tended to be of the same cell immunophenotype. Additionally, genome-wide association studies (GWAS) had already identified SNPs associated with pediatric ALL risk that had a higher frequency in Hispanic/Latino populations. This level of evidence was nowhere near enough to rely on as an explanation though. The known ALL-associated SNPs enriched among Hispanic/Latino-associated failed to explain much of the disparity. This inspired the authors to look for other variants lurking undiscovered.
Thus, the authors carried out their own trans-ancestry GWAS on childhood ALL using 1,878 cases and 8,441 controls from the California Cancer Records Linkage Project (CCRLP). In their results, they identified many SNP associations at the IKZF1 locus, on the short arm of chromosome 7 (7p12.2). This caught their eye. However, GWAS signals are mere tags of specific chunks of DNA associated with a trait. In other words, the variant that gets pulled out of the analysis may not be the variant responsible for the associated trait. There may be another variant buried in the chunk associated with the GWAS hit. Because of this issue, the authors evaluated the GWAS signals carefully with respect to the ancestry of their study population and also carried out what’s called a “fine-mapping” of the region.
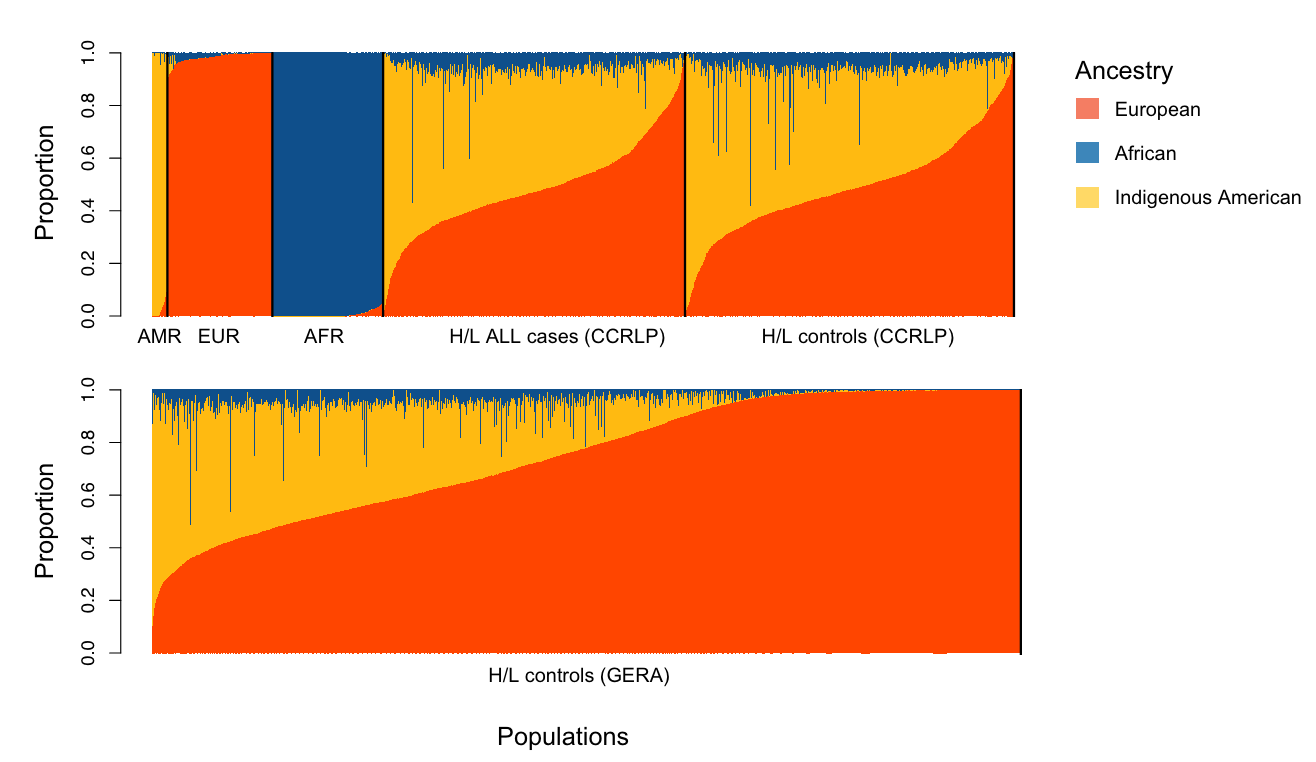
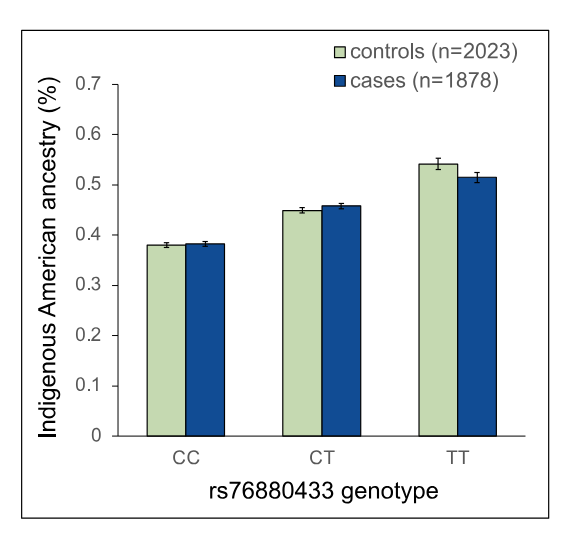
But before the fine-mapping, the authors closely evaluated the three independent, meaning unlinked, GWAS signals prominent in the Hispanic/Latino children: rs4917017 (signal 1), rs10272724 (signal 2), and rs76880433 (signal 3). Taking these together, the authors calculated a genetic risk score at IKZF1 for ALL and found that these three variants may account of 20% of the variation in ALL risk in Hispanics/Latinos while only 6% in non-Hispanic whites. Further comparison with non-Hispanic white individuals highlighted the fact that the signal 3 loci rs76880433 is closely associated with ancestry in Hispanic/Latino population, 30% are carriers versus less than 1% of non-Hispanic whites. This risk loci conferred a greater than 1.4-fold increased odds of developing ALL, interestingly mirroring the discrepancy in incidence, 1.3. Importantly, two of the three risk loci (rs4917017 and rs76880433) were positively correlated with indigenous American ancestry with rs76880433 showing the greatest correlation.
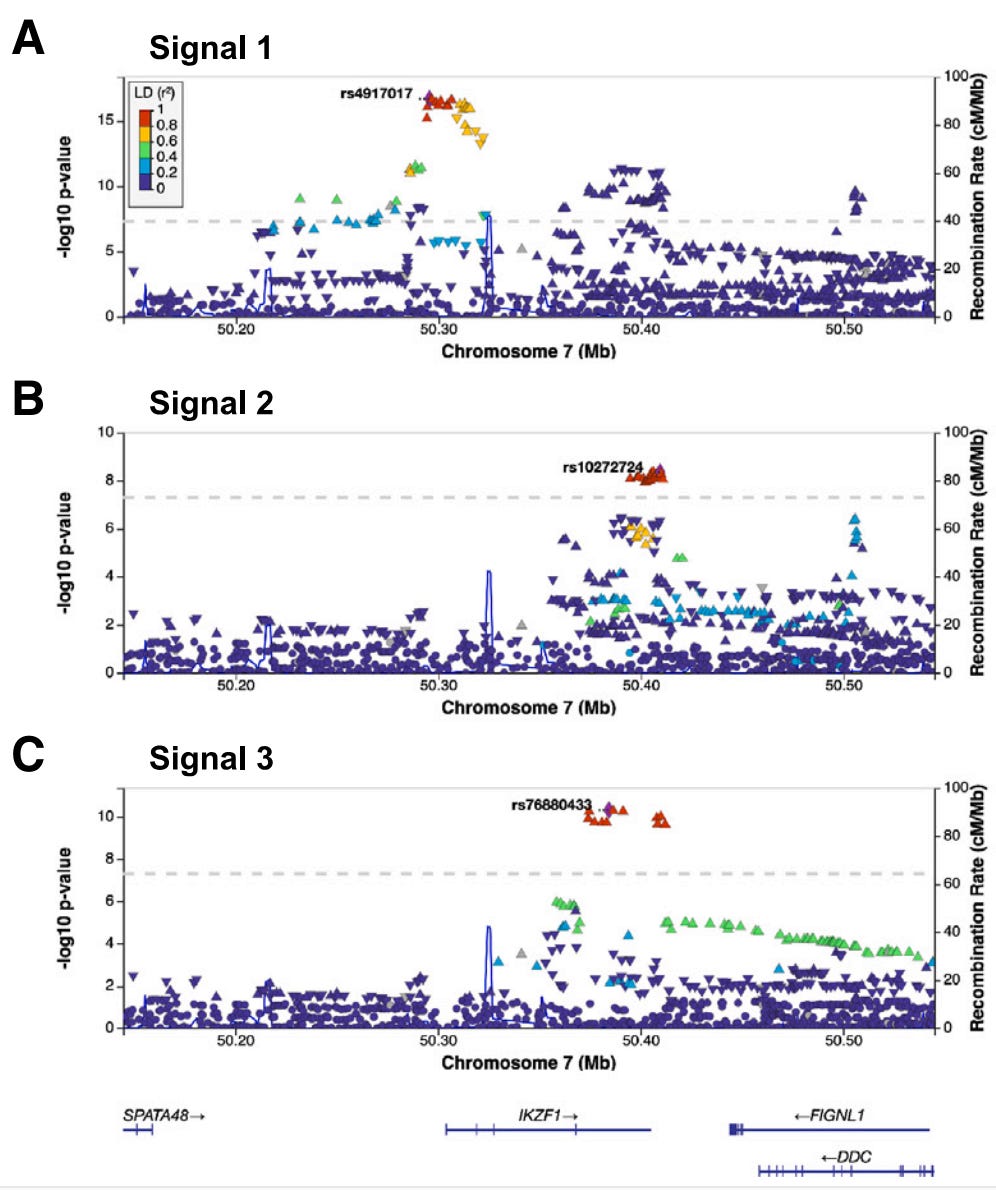
The fine-mapping approach generated three sets of putative causal variants, totaling 44 variants in all. These variants were underlying, as in tied to, the three GWAS signals. There was a particularly interesting variant in the first set. Of the 10 variants in this set all non-randomly associated (linkage disequilibrium) with signal 3, the rs1451367 variant overlapped a DNA region called an enhancer that regulates the expression of IKZF1.6 Further, this rs1451367 variant is in high linkage disequilibrium (LD) with rs76880433 in the Hispanic/Latino population.7 There were some similar variants in the second set with that overlapped with the same region and were just 26 base pairs from the putative causal variant in the first set, rs1451367.
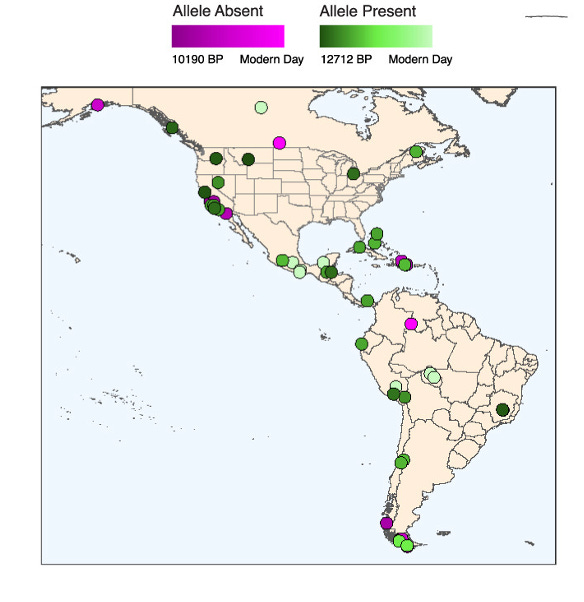
Intriguingly, the rs1451367 is found in the ancient DNA of Anzick-1, a nearly 13,000 year old Paleo-Indian male infant. Anzick’s remains were found in Montana in 1968. Anzick was heterozygous for rs1451367, which supports the claim that many of the first migrants to the Americas also carried this allele. The authors found further support for this in other samples of ancient DNA from the Americas (see figure below).
Given the signs pointing to rs1451367 as the potential culprit behind the increased ALL risk associated with IKZF1 in Hispanic/Latinos, the authors sought evidence of selection at IKZF1. Strikingly, there was some. At the lead SNP for rs1451367, rs76880433 (signal 3), the authors found evidence of selection in Hispanic/Latino populations but not in European (Iberian) or East Asian (Han Chinese) populations.8 A mitigating factor here is that the likely causative variant rs1451367 is estimated to be much older (130-260 kya for rs1451367 versus 28-52 kya for rs76880433). Thus, the authors have greater evidence for selection at rs76880433. No evidence of selection was produced for the other GWAS risk alleles in IKZF1. All in all, the authors have some confidence in and favor the existence of positive selection at IKZF1 in the Indigenous American population despite some of the limitations of the analysis.
One of the more impressive aspects of the paper was the authors explore the likely functional effects in great detail. They move well beyond just showing the GWAS risk variants and the putative causal variant associate with decreased IKZF1 expression in B cells (an eQTL finding). They produce several lines of evidence that indicate rs1451367 contributes causally to ALL risk by affecting IKZF1 in B cells. First, they show that the regions with the risk variants align with regions of chromatin accessibility in B cell and their precursors. They then reasoned that these variants may influence ALL risk by affecting the transition of B cells from progenitor to precursor stages. Leveraging other data from a chromatin capture type experiment on B lymphoblasts, they show that the IKZF1 promoter interacts with the enhancer region harboring rs1451367. Interestingly, the promoter region itself harbors the signal 1 SNP, rs4917017. Together this suggested a three-dimensional interaction of the regulatory regions of IKZF1.
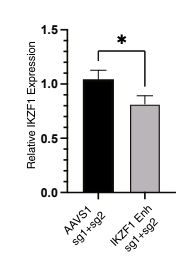
To follow up the in silico evidence, they performed in vitro assays that assessed the regulatory impact of the risk alleles on IKZF1. They showed that the ability of rs1451367 to affect enhancer activity was specific to a progenitor B cell model. They found evidence that IKZF1 bind the enhancer region in which rs1451367 is located and that if they used CRISPR/Cas9 to remove the enhancer region in B cell differentiated primary human hematopoietic stem and progenitor cells (HPSCs), IKZF1 expression was reduced by 20% compared to controls.
This study is not the final word on the increased ALL risk among Hispanics/Latinos, but it does shed light on the intricacies of the molecular and evolutionary mechanisms that can underlie such epidemiological observations. Using a small but diverse dataset along with a slew of other genetic resources, the author identify a single genetic variant that is likely behind a significant portion of this disparity. It is a great case study in the importance of gather clinicogenomic information from as many populations as possible, past and present.
In addition to the increased incidence, young Hispanic patients with ALL have lower overall survival rates even after social determinants of health are accounted for.
Online Mendelian Inheritance in Man (OMIM) and National Comprehensive Cancer Network (NCCN) both recognizes IKZF1 as a known monogenic risk gene for pediatric ALL. The Clinical Genome (ClinGen) resource interestingly has not published a curation of the gene-disease relationship between IKZF1 and ALL.
There is an academic debate about when and how the America’s were populated. The crossing of the Bering strait ~13 kya is the traditional archaeological narrative. However, recent research using ancient DNA methods has complicated this story, extending the early migration deeper into the past. This and other work also suggests seafaring may have been more important than the land bridge in the migration.
Many of the canonical examples of recent selection on traits in different human populations have concerned physical traits like skin pigmentation or metabolic traits like lactase persistence.
The truth will out. Sunlight is the best disinfectant. Fill in whichever cliche about the eventual triumph of truth here. This may make some readers roll their eyes, but over time incontrovertible evidence settles questions. And when questions cannot be resolved by definitive evidence, they are possibly beyond the remit of science. Our duty as scientists is to get to the truth, whatever it may be, as quickly and accurately as possible.
This region was identified as a likely enhancer by the authors in part because the region is located downstream of IKZF1 in a regulatory region defined by characteristic histone modification peaks. They later produce functional evidence to verify the regulatory capacity of the region and the effect of the SNP of interest.
This is a simplification here. The population showing the high LD is AMR or admixed Americans. The r-squared was greater than 0.96 in that population.
There are debates about what level of evidence to accept as evidence for positive selection at some locus in human population. The standard used by the authors is as follows: “The evidence of positive selection is based on the over-representation of lineages carrying the derived allele at the tip of the genealogy, given the distribution of carrier and non-carrier haplotypes when the branch carrying the allele of interest first branched into two sublineages.”
My immediate thought is that this population the United States is repeatedly exposed to huge doses of pesticides over years, as they work in the fields, starting in childhood. And this is more prevelant amongst those with indigenous ancestry as they are the most likely to be impoverished enough to need to work in the fields. And I speak as someone from a family of farmworkers. My Grandmother worked from age 6 to 18 in the fields. Her children didn't have to. Some never make it out.
A stupid question, if you don't mind - does "1.3-fold" this mean 30% higher or 130%? (2 fold is doubling, so 1.3 fold is 30%?)
and also, does the paper go into whether IKZF1 could have originated in Asia and then come over with the earliest migrants to the the Americas? Was the evidence for selection rather than founder effect/bottleneck?