

Why Do BRCA Mutations Cause Breast Cancer?
Why Do BRCA Mutations Cause Breast Cancer?
Connecting BRCA gene mutations to hereditary breast and ovarian cancer was just the beginning. Precision medicine is supported by discovering mechanisms.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
An important point that my previous post on inherited breast cancer glossed over was that mutations in breast cancer risk genes often confer cancer risk upon other organs than just the breast. I did mention ovarian cancer without providing much background, but it was hard to leave out since germline mutations in BRCA1/2 qualify as a diagnosis of hereditary breast and ovarian cancer (HBOC). In this latest edition to Cancer Genomes, we’ll explore organotropism,1 differential clinical presentations, and variant effects in BRCA-related HBOC.

Why Do Mutations in Cancer-risk Genes Confer Risks to Particular Organs?
In the context of cancer, organotropism typically refers to the propensity of certain metastatic cancer cells to colonize certain organs. A simple metaphor for the idea of orgnanotropism is that of seeds that will grow in some soils but not in others. For instance, prostate cancer patients are often afflicted by metastasis to bone but not to the brain. Organotropism is an important phenomenon in cancer that is studied actively. However, in the context of inherited cancer, I’d like to tweak this definition of organotropism a bit to refer to the propensity of mutations in certain genes to associate with cancer in specific organs.2 In our prior post, we discussed how BRCA1 mutations confer a high risk for cancer of the breast and/or ovaries in families. The natural questions this prompts: Why are those the organs affected and not others?3
There are two simple and unsatisfactory answers to this question:
The pattern of organotropism observed in HBOC or other cancer syndromes is a function of the complex interactions among the inherited mutation, the genetic background of the mutation carrier, the default cellular physiology of the at-risk organs, and the exposures experienced by the carrier. Presumably, there is some sort of relationship between the mutant gene and the default functions of the cells in the at-risk tissue.
We have no detailed idea concerning what explains the flavor of genetic organotropism observed.
Now, that I’ve concede that we know everything about how organotropism must be mediated yet know nothing with confidence. I will treat readers to a more detailed look at the possible biology that may shed some light on why mutations in BRCA genes tend to cause cancers of the breast and thyroid.
Tissue specificity of gene expression
Some genes are expressed only or mainly in certain tissues or organs. Subsequently, mutations in tissue-enriched/specific genes are likely to affect the function of the cells in those tissues or organs. For example, the gene PAX8 is a transcription factor that regulates the development and differentiation of kidney cells. Genetic variation that affects PAX8 can help drive kidney cancer by activating oncogenic signaling pathways.
Is this tissue-specific story what’s going on for the BRCA genes? The answer appears to be no. Analyses that have looked at BRCA1/2 expression across tissues at multiple biological levels (RNA and protein) show a broad pattern of expression across tissues and no special enrichment in the breast or ovaries.
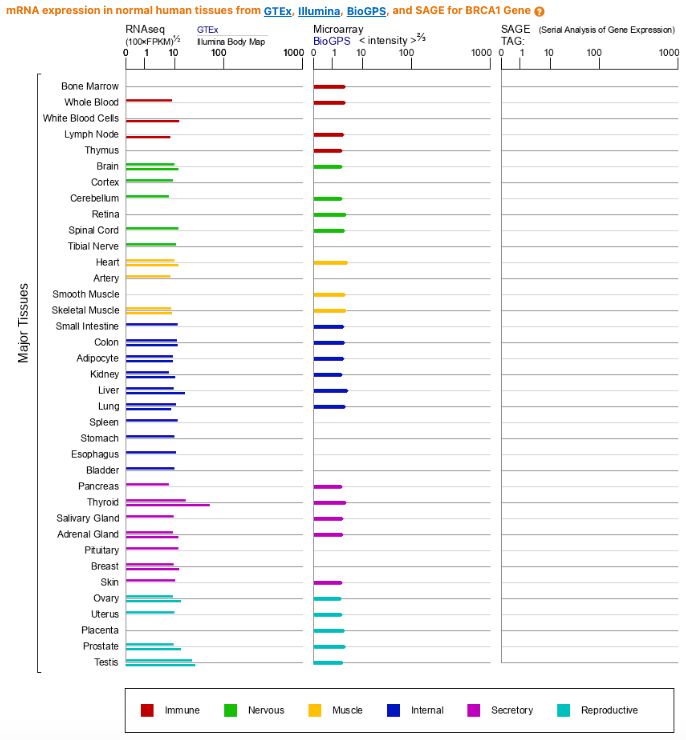

Tissue specificity of mutation exposure
Some tissues or organs are more exposed to environmental or endogenous factors that can cause mutations or drive tumorigenesis: ultraviolet radiation, tobacco smoke, and steroid hormones. These factors tend to increase cancer risk by either damaging the DNA of cells or by signaling that cells should grow and proliferate when they shouldn’t. The relevant exposure in HBOC is estrogen (and other sex hormones to a lesser extent). Despite the likelihood that this connection bears fruit, the literature on the subject is unsatisfying. There are some tantalizing hints, but also some suggestions that it is a dead-end. For instance, there is no increased risk of breast cancer for BRCA1 mutant carriers in menopause who received hormone-replacement therapy versus those who didn’t. The pattern of organotropism in HBOC strongly suggests a connection between endocrine physiology and BRCA gene function, but the exact nature of this relationship remains ambiguous.
Tissue specificity of mutation selection
Some tissues or organs have a higher rate of cell division or turnover, which can increase the chance of acquiring mutations during DNA replication. These mutations can confer a selective advantage to the cells by enhancing their growth, survival, or resistance to stress. For example, the lining of the colon has a high rate of cell turnover, which can lead to the accumulation of somatic mutations. Often times, the the gene APC is affected.4 Mutations in APC further potentiate this process of increasing genomic instability and uncontrolled cell division eventually transforming into colon cancer.
Both the breast and ovaries are dynamic tissues where cellular proliferation occurs. However, this observation prompts the question: why aren’t other proliferating tissues sprouting cancers in BRCA mutation carriers? Or at least why don’t other proliferative tissues show as high of a risk as the breast and ovaries do? I make this qualification as there is some evidence to suggest that BRCA carriers do have higher cancer risk in other tissue like the colon. So like the hormonal exposure, the proliferative nature of certain tissues bears some fruit too but provides little in the way of specific insights into the observed pattern of organotropism.
Tissue specificity of genetic interactions
Increasingly, there are hints of a composite genetic risk profile for certain traits. Say you inherited a BRCA1 mutation and with it a genome that also confers additional background risk for breast cancer. If you are in this unfortunate situation, it does appear you would be at higher risk than an alternate hypothetical person who inherited the same BRCA1 mutation and a genome of low overall risk for breast cancer. This scenario gets at the idea of genetic interaction or a composite genetic risk profile that factors rare and common variation into to the total likelihood of presenting with a given cancer. Although this formulation provides more diagnostic insight, it is only obliquely related to organotropism.
Given the enduring mystery of organotropism in BRCA mutation carriers, it is worth comparing how the BRCA1 and BRCA2 versions of HBOC compare clinically. The comparison may be revelatory and at the very least it is the beginning of establishing a reliable and mechanistic understanding of genotype-phenotype relationships.
Clinical Perspective: Differences Between BRCA1- and BRCA2-Associated HBOC
There are subtle clinical trends that distinguish BRCA1-HBOC from BRCA2-HBOC. First, BRCA1 mutations are more strongly associated with cases of hormone receptor (HR)-negative cancers or triple-negative breast cancers, while5 BRCA2 mutations are more associated with HR-positive breast cancers. Further, BRCA1 mutations are associated with a higher risk of ovarian cancer (39%-58% lifetime risk) than BRCA2 mutations (13%-29% lifetime risk). However, men with BRCA2 mutations have a higher risk of breast cancer (1.8%-7.1% lifetime risk) than men with BRCA1 mutations (0.2%-1.2% lifetime risk). Both BRCA1 and BRCA2 mutations are associated with increased lifetime risks of pancreatic cancer (5-10% for BRCA1/2) and prostate cancer (7%-26% for BRCA1 and 19%-61% for BRCA2) in carriers.6 Both pancreatic and prostate cancers should be considered part of HBOC’s larger spectrum of organ-specific cancers, though confidence in the relationship is not quite as strong. Both are endocrine tissues so in some ways they are a natural expansion to the already known organ-specific risks.
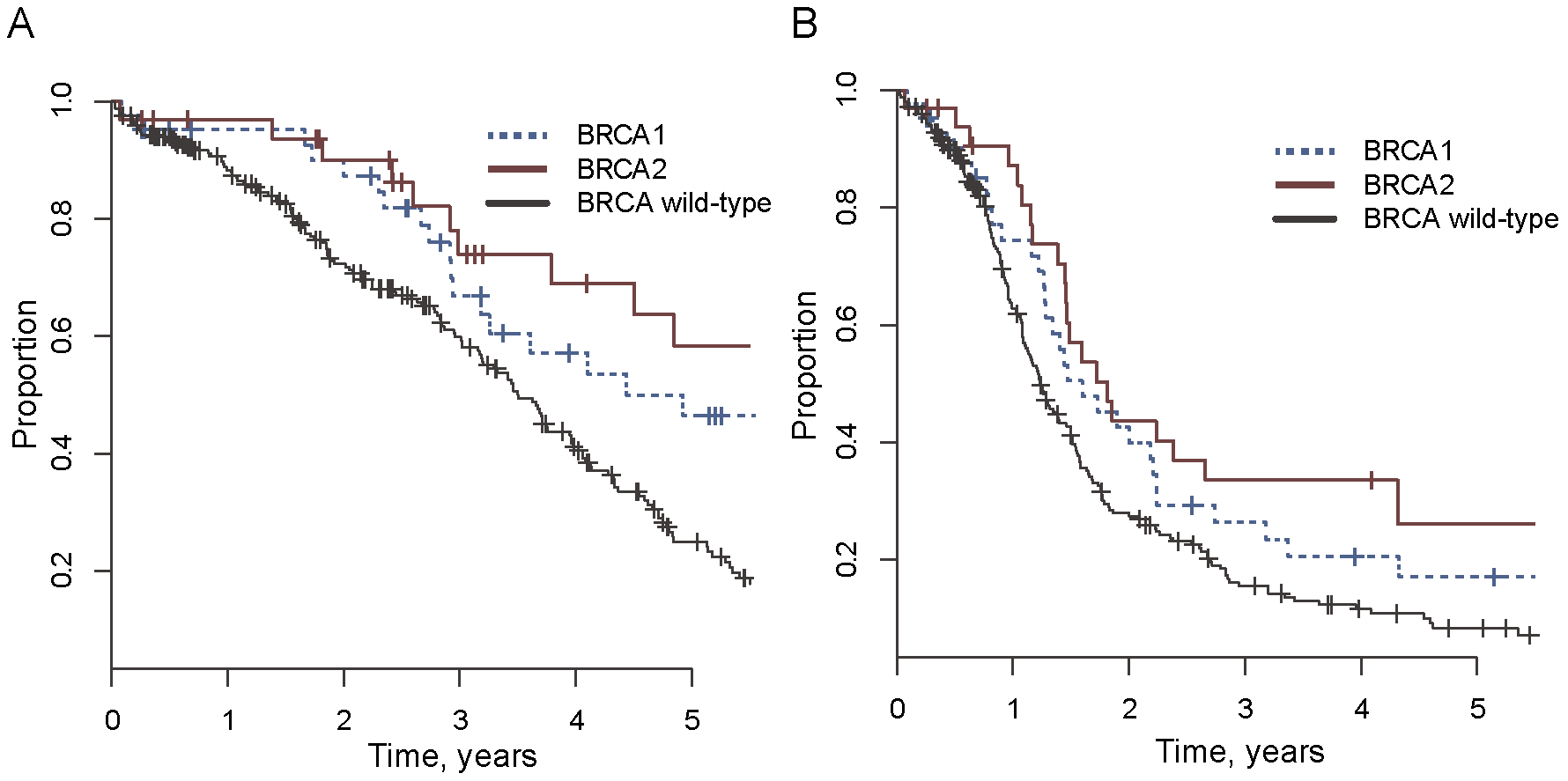
Generally, this brief comparison of clinical presentations probably suggests to readers that BRCA1 mutations are associated with more aggressive disease and worse outcomes. This is in fact true. Survival rates tend to be lower among BRCA1 mutation carriers across the board (the figure above compares survival across BRCA status in ovarian cancer). The greater severity of BRCA1-HBOC is probably explained by the higher rate of triple-negative disease and a higher risk of ovarian cancer, which has lower survival rates than breast cancer.7
Given that significant mortality risk is associated with BRCA mutation, early diagnosis, especially before developing cancer is critical. Having an earlier genetic diagnosis enables proactive clinical management. This can include regular cancer screenings and prophylactic surgeries, such as the removal of currently healthy breast and ovarian tissue. Currently, the National Comprehensive Cancer Network (NCCN) recommends a risk-reducing salpingo-oophorectomy (RRSO), the removal of the ovaries and fallopian tubes, between 35-40 years-of-age for BRCA1 carriers and between 40-45 years-of-age for BRCA2 carriers.8 Risk-reducing mastectomy (RRM), the removal of the breast(s), does not have a blanket recommendation from NCCN but are often encouraged in high-risk carriers. According to a 2010 study, the clinical action associated with the highest survival probability (i.e. the probability for a current 25 year-old BRCA1 or BRCA2 carrier to reach 70) is RRSO at age 40 and RRM at age 25, though there are often marginal differences between many different strategies (see figure below).

All things considered, the clinical management of BRCA1/2-HBOC looks good. Getting an early genetic diagnosis is key,9 but once that is obtained then following recommended clinical management protocols with an appropriate specialist appears to nearly guarantee a normal lifespan. Individuals who get risk-reducing prophylactic surgeries have taken to calling themselves previvors. The empirical data appear to support such labeling! Of course, there is still more to learn, and I hope we see more large, prospectively accrued sequencing projects yoked to careful and continuous phenotyping. These will provide ever greater confidence and resolution to our clinical recommendations, and they will enable more comprehensive understandings of genotype-phenotype outcomes. This will help us make continued strides in precision medicine.
A Peek at Understandings of Variant Effect in BRCA1/2
Interpretation of genetic data is a major hurdle. It requires a lot of rigorous work. Fortunately, a significant amount of this hard work has been done by academic and industry scientists for BRCA. Much of the variant classification data was private though, but it is now being shared publicly by Myriad Genetics. This was the right move on Myriad’s part in terms of prioritizing patient outcomes generally, though critics of Myriad say this is too late in the game to be praised. Regardless, even this new data doesn’t entirely solve the interpretation problem, especially because new variants are being identified everyday. In these cases, we have to turn to research on variant effects.
In 2018, the first large-scale multiplexed assay of variant effect (MAVE)10 was published for BRCA1. The study linked saturation mutagenesis of 13 high priority BRCA1 exons to a functional assay using cells dependent on the introduction of a functional BRCA1 protein for survival. Subsequently, functional effects were able to be measured and reported for roughly 4,000 single nucleotide variants (SNVs), and the effects were observed to be bimodally distributed, a clump of benign and a clump of damaging variants. The derived functional score were nearly perfectly correlated with established assessments of pathogenicity too, meaning the assay was likely reporting clinical meaningful information about variant effect. Finally, about 300 new SNVs that disrupt BRCA1 expression were found. MAVE studies like this hold the promise to completely resolve variant interpretation during genetic analysis of cancer risk genes. They are an important part of the future of clinical genetic, though have yet to really touch the practice of clinical genetics.

Cancer Genome Futures
This wraps up BRCA1/2 and HBOC. Please send questions and feedback. I do feel this was a somewhat brief treatment of BRCA1/2. The series on hereditary cancer will continue with a look at the guardian of the genome, TP53. Stay tuned!
Organotropism describes the affinity of drugs, pathogens, or metastatic tumors for specific organs, organ systems, or somatic tissues. Metastatic organotropism is the non-random process of distant metastases distributing to certain organs. In this post, I will offer a bit of a tweaked definition on organotropism in the context of inherited cancer risk.
I think my usage still leverages the ideas embedded in the base work “trop” meaning “to plant to turn” in Greek. My usage harkens back to Charles Darwin’s nascent conception of genes as “gemmules,” from the Latin for “bud,” in his theory of pangenesis. It is still suggestive of the seed-soil metaphor I mention when defining organotropism.
As more and more genome sequencing of suspected inherited/familial cancer cases is performed we have been learning that the organ-specificity associated with mutations in certain genes is looser than previously thought. For instance, we now know that BRCA1 mutations also confer risk for pancreatic cancer on top of the breast and ovarian risk.
A gene we’ll return to in a later addition of the Cancer Genomes series.
Triple-negative breast cancer is a subtype of breast cancer that is particularly aggressive and is notable for its lack of expression of the estrogen receptor, progesterone receptor, and a receptor-tyrosine kinase called HER2.
Lifetime risk figures for organ-specific cancers in BRCA1/2 carriers was reported from the latest NCCN guidelines on breast cancers.
The 5-year survival rate is 90% for breast cancer but around 50% for ovarian cancer.
BRCA2 mutation carriers can delay RRSO because onset of disease is typically later. This is another way in which BRCA1 disease is more aggressive.
Genetic testing is often indicated for individuals with a first-degree family member with a confirmed pathogenic/likely pathogenic mutation, family history of breast or ovarian cancers, Ashkenazi Jewish ancestry, or specific cancer histologies like triple-negative disease. Please consult a medical professional if you have questions about whether genetic testing is appropriate.
Recall my prior post on MAVE studies. Please return to it, if a refresher is needed.