

Kidney Anatomy as a Metaphor for Wilms Tumor Genetics
Kidney Anatomy as a Metaphor for Wilms Tumor Genetics
Cancer biology and organ development intersect in a circuitous tangle in the kidney.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
tl;dr section
Wilms tumor (WT) is a pediatric cancer of the kidney with generally favorable outcomes to surgery plus chemotherapy
The genetic study of WT implicates many genes at the nexus of kidney development and cancer growth. Much remains to be learned about both process by studying WT. Heterogeneity in WT remains a hurdle to such studies.
Precision approaches beyond DNA sequencing for diagnosis, such as patient-derived xenograft models and liquid biopsies may hold promise for improving care.
In 1899, a young German surgeon Dr. Max Wilms penned "Die Mischgeschwulste Der Niere" or “The Mixed Tumors of the Kidney.” This is the first known description of nephroblastoma or what has become eponymously called Wilms Tumor (WT). “Mixed” is certainly an apt word here. As not only is nephroblastoma histology mixed, but the genetic architecture is too. It is a mess of overlapping and intersecting biology at the nexus of cancer and development. It is reminiscent of the kidney’s complex arrangement of functional units, nephrons, or the squiggliness of the nephron itself.
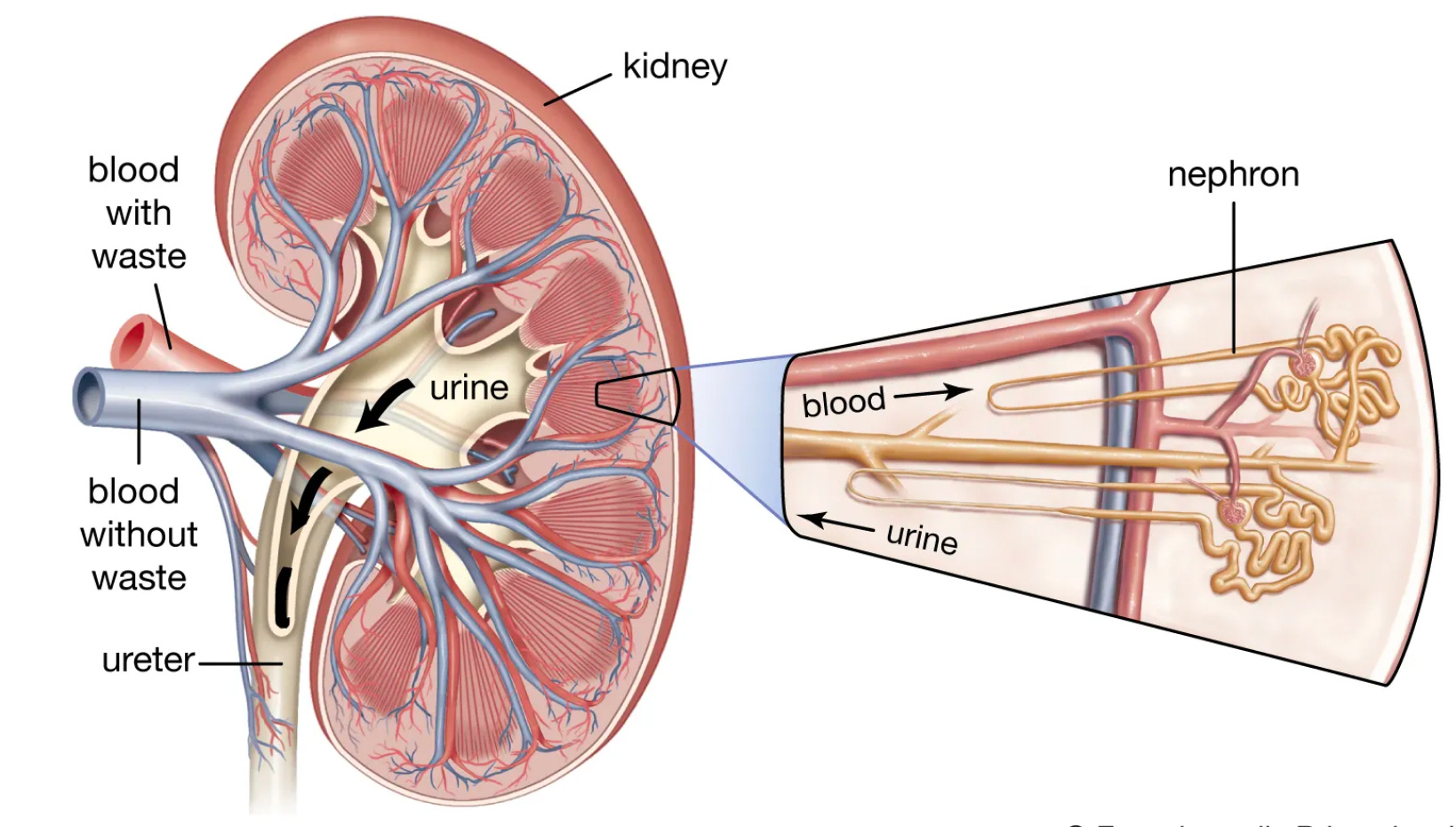
Unfortunately, Dr. Wilms’ life was cut short by septic shock, which appears to have been the sequelae of an infection he picked up performing an operation on a prisoner in WWI. Nonetheless, his careful pathological work birthed an entity that trained the focus of biomedical science on a deadly phenotype at the “intersection between disrupted organogenesis and tumorigenesis.” A comprehensive dissection of the tangled genetic architecture may therefore shed light on the shared and distinct biology of these processes.

Wilms Tumor
Wilms tumor (WT) stands as the most common kidney cancer in children, affecting 1 in 10,000. Across the globe, this computes to roughly ~14,000 annual diagnoses and roughly 5,000 deaths, many of which are preventable.1 WT is thought to arise during embryonal development. Consistent with this model, it frequently arises within or in association with “nephrogenic rests,” islands of remnant embryonic renal tissue within an otherwise mature kidney. Given the embryonal nature of WT, it is unsurprising that the median age of onset is quite low, at 3.5 years-of-age. For reasons unknown, WT is somewhat more common in girls - a difference that usually goes the other way in childhood cancers. There is also some variation in incidence among different ancestry groups. For example, in the United States, African Americans have a nearly three times higher incidence than Asian-Pacific Islanders. The genetic heterogeneity of the disease is thought to contribute to the ancestral differences.



Altogether kidney cancers represent 5% of malignancies before 15 years-old.2 Thus, it has become an important model of pediatric malignancy that science has relied on to gradually learn how to optimize care. WT presents both as genetic disease and sporadically. It can present with a single lesion or multiple unilateral lesions or bilateral lesions. Even in inherited cases, the genetic picture is still complicated relative to other inherited cancers. There are roughy 40 cancer-risk genes implicated in WT predisposition. The cause of sporadic cases are more ambiguous, but it is still hypothesized that the same biological pathways are involved. It is often thought, especially for sporadic bilateral tumors, that early post-zygotic founder mutations occur in the usual risk genes in somatic cells of the developing kidney.
If possible, WTs are treated with surgery. Surgical resection is followed by chemotherapy; this is referred to as adjuvant therapy. Sometimes chemotherapy is given in the neoadjuvant setting (before surgery) too. Regardless, the outcomes with either treatment approach are good. Overall survival rates are now above 90%. These high survival rates have been primarily achieved by adopting a risk-guided approach to treatment and using an effective two-drug cocktail, vincristine and actinomycin D. The first drug is a natural alkaloid isolated from the plant Catharanthus roseus, which goes by many common names including Madagascar periwinkle.3 Vincristine binds irreversibly to microtubules, important cellular structures, and arrest cell division (mitosis). The second drug was the first antineoplastic antibiotic, i.e. a drug that can kill bacteria and tumor cells, isolated from the bacterium Streptomyces parvulus. Actinomycin D binds DNA and disrupts transcription of DNA to RNA and DNA replication.
Unfortunately, this treatment approach (surgery & chemotherapy) is not perfect. 20% of cases will relapse after treatment and a quarter of survivors will have significant morbidity subsequent to treatment. There is active research to address the long-term issues created by radical nephrectomies, including a closer look at nephron-sparing surgery (NSS). Additionally, next-generation sequencing (NGS) approaches are being used to find ways of targeting disease-relevant pathways and/or guiding treatment.
A Genetic View of Wilms Tumor
It is clear that genes involved in the development of the kidney (nephrogenesis) are liable to be risk factors for WT. Cancer and development are obviously separate, complex processes, but the overall genomic program is often efficient in repurposing genes contextually in variably ways. There are also redundancies, where different genes provide input on the same processes. So the picture is less of a nice lattice of intersections and more of a tangle of reticulation. Obviously, many genes are thus implicated in germline risk. There is also some slight variation among experts in reporting these predisposition genes. Filippo Spreafico and colleagues argue the primary predisposition genes WT include WT1, CTNNB1, and WTX (AMER1). They also include loss of H19-IGF2 imprinting (11p15) as an important epigenetic risk factor.4 Alternatively, Joyce Turner, Jack Brzezinski, and Jeffrey Dome highlight five major loci associated with WT predisposition: REST, TRIM28, WT1, 11p13, and 11p15. Both groups highlight 20 or more other genes that contribute in a minor way to WT predisposition. These other genes are often involved in more neatly delimited cancer syndromes where WT risk is a lumped in phenotype at some risk level (usually low).

Because the germline risk landscape is so vast. The focus of research has been more interested in identifying the central contributors to Wilms tumorigenesis (the putative landscape of central operatives is shown below). This is ongoing work as modeling the disease in animals has been difficult and even next-generation approaches cannot completely unlock the mysteries created by the combination of rarity and heterogeneity. However, there is hope that singe-cell whole genome and multiomic analyses may resolve some of the heterogeneity. An additional challenge here is that treatment without a precision approach is already quite good, which has led to more strategic positioning of resources and attention beyond solely molecular analysis of WT.

As mentioned above, a good indicator of the genetic heterogeneity underlying the risk profile of WT is that it pops up across many different genetic syndromes. Almost 20%, of WTs occur in the context of genetic syndromes. This prominently includes WAGR syndrome and Beckwith-Wiedmann syndrome (BWS). WAGR Syndrome is named for its key features: Wilms tumor (W), aniridia (A),5 genitourinary problems (G), and intellectual disability (R). Study of WAGR helped connect the loss of the gene WT1 (11p13) to WT events. This appears to be the closest thing WT has to its own syndromic diagnostic entity. BWS cases display general overgrowth, often noticeable at birth, alongside an increased propensity for several pediatric cancers, including WT. Intriguingly, imprinting, an epigenetic phenomenon where a gene's activity depends on whether it's inherited maternally or paternally, plays a significant role in BWS. The study of BWS cases connected H19-IGF2 imprinting to WT events.

There is research interest in the emerging genetic factors involved in WT etiology and tumorigenesis. These insights have been derived by study of WT tissue and thus variably figure into the predisposition equation. Rather than diagnosis alone, the more pressing aim is to refine prognosis and treatment targets and strategies. Some of the interest in driven by some of the relative novelty of the pathways at play. For instance, the biogenesis of microRNAs (miRNAs) has recently drawn attention. Mutations in microprocessor genes DROSHA and DGCR8 are common in WT cases. The latter is predominantly observed in female cases of WT, which is an intriguing, unexplained tidbit. The RNAse encoded by DICER1 has also been found mutated in WT in rare cases and has also been linked to kidney sarcomas. These lines of study have enabled gene-disease histology association to be developed as illustrated below.
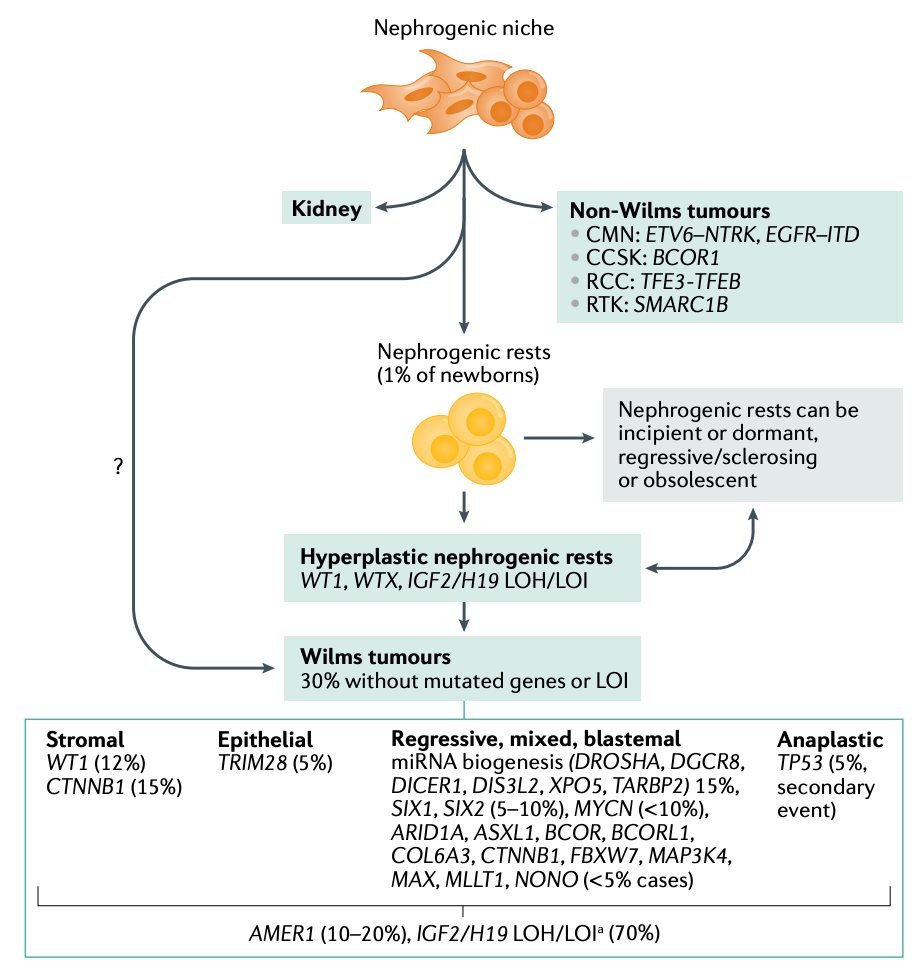
Advances in Precision Approaches to WT
Drug screens performed with patient-derived xenografts (PDXs) have driven novel target identification in WT. PDXs are preclinical cancer models created by directly implanting a patient's tumor tissue or cells into an immunodeficient mouse. The mouse host then acts as a surrogate environment for the cancer to grow and evolve. Unlike traditional cell lines, PDXs retain a significantly higher degree of similarity to the original patient tumor. They preserve the tumor's diverse cell populations (heterogeneity), the three-dimensional structure, and more closely mirror the genetic and molecular complexity of the primary tumor. By treating PDX models with potential drug candidates, scientists can find new drug targets, identify pathways essential for tumor progression and survival, and evaluate how mutations influence responses to targeted therapies. The insights derived are considered more translatable to the bedside.
In WT, there are a number of ongoing investigational effort powered by PDX-based findings:
The ADVL0821 phase 2 trial (NCT00831844) of cixutumumab, an antibody directed against the human insulin-like growth factor-1 receptor (IGF-1R).
No response reported.
The ADVL1121 phase 2 trial (NCT01502410) of sorafenib, a small molecule multikinase inhibitor that blocks the effect of angiogenic cytokines.
No response reported.
The ADVL1622 phase 2 trial (NCT02867592) of cabozantinib, another small molecule multikinase inhibitor that blocks vessel growth.
No response reported.
The ADVL0921 phase 1 trial (NCT02444884) of alisertib, a small molecule Aurora A kinase inhibitor.
Complete Response in one WT patient reported.
The phase 2 trial (NCT00331643) of ixabepilone, an antimicrotubule agent.
Two prolonged stable responses reported.
The ADVL1522 phase 2 study (NCT02452554) of lorvotuzumab mertansine, an antibody-drug conjugate comprised of a CD56-targeting antibody in conjugation with the cytotoxic maytansinoid DM1.
No response reported.
The phase 2 study (NCT04851119) of tegavivint, a small molecule inhibitor of the Wnt/beta-catenin pathway.
This trial is actively recruiting.
Although some of these studies have yet to complete, the overall response data is clearly not particularly positive. In my quick survey of the trial results, only one WT case has shown a complete response to any of these drug, alisertib. Nonetheless, the PDX approach will certainly be useful in a disease like WT and can be potentially utilized in real-time to guide patient treatment.
In addition, WT may be amenable to liquid biopsy (LBx) approaches for diagnosis, treatment response, and minimal residual disease (MRD) detection. LBx is a minimally invasive method to sample and analyze tumor-derived components from bodily fluids, primarily blood, typically using NGS technologies. It targets components like circulating tumor cells (CTCs), circulating tumor DNA (ctDNA), and exosomes. The Children’s Oncology Group (COG) study AREN1921 (NCT04322318) is currently collecting serial blood and urine samples from newly diagnosed anaplastic WT cases or from relapsed favorable histology WT cases. The results of this study will indicate whether LBx holds promise in WT.
Rare Disease Requires Cooperation and Creativity
WT is a currently a good illustration of how cooperative and innovative efforts among investigators and medical professionals can speed up research and improve outcomes. The COG and the International Society of Pediatric Oncology (SIOP) have organized prospective trials that sync clinical and biological aims, including strategies like biobanking and risk stratification studies. Plus, these independent group efforts enable cross-validation via meta-analysis, which is crucial when working with small groups of rare patients. This work along with broad and standardized sharing of data, especially of the molecular variety, will catalyze innovation. WT may not be the best example case for using genetics to drive precision care, but it is a disease that may offer return on investment for the field. If we can dissect the complex biology, tease out the intersecting processes of cancer and development, we may unlock translatable insights and amplify the impact of precision approaches.

Most of the mortality associated with Wilms tumors (WT) is related to regional variation in treatment availability and quality. Thus, low-income areas see significantly more deaths from WT.
Essentially all of these are WT.
It is endemic to Madagascar.
This “imprinting” language essentially refers to the configuration of how the H19-IGH2 (11p15,5) region of the genome is inherited from the WT patient’s mother. The changes to the maternal allele from hypermethylation cause the IGF2 gene to be overexpressed. IGF2 is a growth factor.
An abnormality of the iris where it seems absent.