

A Vision of Cancer's Past
A Vision of Cancer's Past
The retinoblastoma story.

This article is part of a series on hereditary cancer syndromes and cancer genetics called Cancer Genomes. If new to the series, please go to my post “Introducing Cancer Genomes” for an explainer.
The wages of cancer is suffering, but insights from genetics offer recourse.
Retinoblastoma is a tragic disease. This rare type of cancer affects the retina of the eye(s) of the young (1:15,000 to 1:20,000 births). Typically, observed in infants but sometimes diagnosed later in early childhood (usually by 3 or by 5 at latest), it can mean a lifetime of impaired vision and a life evading the claws of cancer.1 Although survival rates are excellent, the emotional toll on patients and their parents is hard to overstate.

The Long History of Retinoblastoma
Intriguingly, we potentially have a relatively deep historical record of the disease. The 16th-century Dutch anatomist Pieter Pauw left notes on an autopsy of a 3-year-old boy with an enormous orbital tumor. He performed that autopsy in 1597!2 This gruesome history was exhumed by Julius Hirschberg, a German ophthalmologist and historian, in the 19th century and then popularized in connection with retinoblastoma by the ophthalmologist Edwin B. Dunphy. In a lecture in 1963, Dunphy quoted a translation of Pauw’s autopsy notes almost in full and many others ran with this interpretation.

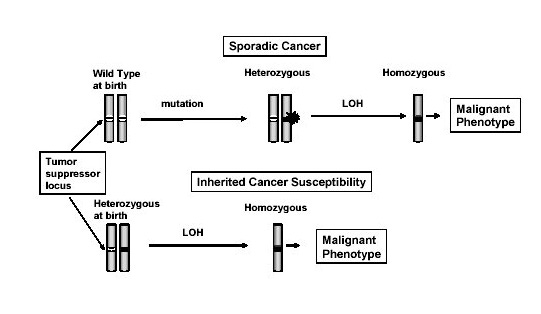
The narrative of retinoblastoma’s long history was emerging in medical consciousness essentially in tandem with observations that suggested it was likely a genetic disease. In 1958, Sheila Maynard Smith and Arnold Sorsby concluded that bilateral (both eyes) cases of retinoblastoma are most often familial, while unilateral (one eye) are likely sporadic. This observation turned out to be essentially correct but somewhat misleading. All cases have a genetic basis - sometimes referred to as constitutional, however, the origin of the causative mutation varies, typically in correlation with whether the presentation is bilateral or unilaterl. The bilateral cases tend to occur when transmission of the mutation intergeneration, from parent to offspring, while the unilateral cases are often new mutations (de novo) that occur spontaneously in the parent’s sperm or egg or sometime during development, either in utero or after.3

These early observations about the pattern of disease (unilateral vs bilateral disease) inspired debate about how to model the data. In 1971, Alfred Knudson presented the 2-hit model, where an individual inherits or already has a mutation in a disease-causing gene and then another mutation in the other copy (allele) of the gene occurs, triggering disease. Individuals with germline mutations would subsequently have higher bilateral risk, while those without germline mutations would have higher unilateral risk of disease because the second hit would be unlikely to occur at the developmental time where both retina would be affected. Ei Matsunaga offered a soft attack on the 2-hit model, claiming it was too improbable to have a repeated second hit in somatic tissue. However, shortly after this contention was raised the mathematical predictions of the 2-hit model were shown to be consistent with the patterns of disease in retinoblastoma pedigrees under a plausible rate of mutation. Plus, the genetic origins of retinoblastoma were discovered, which eventually provided empirical evidence of the phenomenon. RB1 thus represents one of the earliest and most important success stories in cancer genetics. Furthermore, the 2-hit model continues to inform how cancer biologists think about the pathogenesis of hereditary cancer today.
Piecemeal Discovery of RB1, The Retinoblastoma Gene
It is no longer in vogue to name genes after their phenotypes. Many geneticists are eager to sideline essentializing conceptions of gene function. Also, it gets hairy when we call a gene “disease X gene” and then we later learn it is involved with trait Y and Z too.4 However, RB1 was discovered in the pioneer era of genetics so its name is derived directly from its associated phenotype, Retinoblastoma 1. It is designated the first tumor suppressor gene, the genetic “brake pedal” slowing cell division, to be discovered.
In a few of the prior cancer genes I’ve touched on so far, many were discovered using linkage analysis. This is an approach that relies on tracking the mixing and matching of different chromosomal segments with polymorphic markers in association with a trait across generations of a family. Linkage analysis was part of RB1 discovery but wasn’t decisive. Finding the gene responsible for retinoblastoma incidence just wasn’t optimal for linkage analysis because a minority of cases presented in large pedigrees unlike the cancer syndromes with onsets in early middle life. Plus, a lot of the discovery work for retinoblastoma came almost a decade before linkage analysis would mature as a method in the toolkit of human geneticists. These scientists had to rely on some serendipity and get creative with their methods to find RB1. It took the better part of the 1980s to figure everything out, but they got to the truth despite some confusion along the way.
Given the early onset and sometimes clearly genetic pattern of retinoblastoma, scientists took a peak at the chromosomes of patients and often found the loss of a segment on the long are of chromosome 13, specifically 13q14. They also noticed that this was often accompanied a loss or decline in the expression of esterase D (ESD), a gene already known to be in that region. They even produced some low resolution evidence from linkage analysis to allege a relationship between ESD and retinoblastoma. However, ESD was a genetic red herring. It was simply a passenger to the deletions of 13q14 due to its location in the region and close proximity to the actual perpetrator, RB1.
After closer study, scientists were able to hone in on RB1’s location in the genome. A number of different approaches were used. Many of which were similar to the hybridization, cloning, and sequencing approach that was used to discover mutations in TP53 in association with Li-Fraumeni syndrome. These careful studies disentangled the esterase D expression biomarker from the loss of 13q14, identified the RB1 locus, mapped the boundaries of the gene, cloned the gene, found mutations in association with retinoblastoma, and started to empirically substantiated the 2-hit model. Looking back at the literature, it is hard to point to a definitive moment where RB1 appeared as a full fledged gene. It came in many pieces parts throughout the 1980s, where important milestones were reached in 1983, 1986, and 1989.5 By the close of the decade into the 1990s, actual efforts to screen the RB1 gene for mutations in retinoblastoma patients in the clinic were underway. RB1 was now indubitably the risk gene responsible for retinoblastoma, a discovery effort almost 400 years in the making.6
Today, we know that loss-of-function RB1 mutations, mostly nonsense, splicing or frameshift type, are responsible for almost all (~98%) cases of retinoblastoma. However, in most cases the mutation in RB1 occurs spontaneously in one of the parent’s sperm or egg (de novo mutation), during early development of the child (mosaicism), or after development (somatic mutation). Around 2% of retinoblastoma, exclusively unilateral cases without family histories, show alterations in another gene, MYCN. And unsurprisingly, the amplification of MYCN drives retinoblastoma by interfering with RB1 function.

What Does RB1 Do in the Cell?
After the long road to discovery was trod, it was time to explore what RB1 was actually doing and find out why its loss led to retinoblastoma. In the mid-90s, several studies demonstrated that RB1 interacted with a family of proteins called E2F. E2F proteins are transcription factors, meaning they bind to regulatory regions of the genome and influence the expression of genes. RB1 is a repressor of E2F activity. RB1 restrains E2F, preventing it from driving the expression of pro-growth and division genes.7 When cells need to divide a protein complex called CDK4-Cyclin D modify RB1, releasing E2F from its repression.8 E2F then stimulates cells, via changes in gene expression, to enter a critical step in cell division, S phase. S phase is when a cell duplicates its genetic material in preparation to divide into two daughter cells. The division process is irreversible after committing to S phase, making the checkpoint (G1-to-S phase) that RB1 helps regulate especially important to keeping cells under normal genetic control.

Unfortunately, the exact reason why retinal tissue is specifically susceptible to disruption of RB1 is not fully understood.9 However, one can imagine some possible low resolution explanations. Such hypotheses merely draft off of our basic knowledge about developmental processes, mechanisms of cell proliferation, and differences between tissues types. We know that the retina is a highly proliferative tissue during development, and that this process must require exquisite regulation of differentiation and cell cycle processes. So RB1 may be simply be critical to the regulation of unknown factors (in addition to the loss of E2F regulation) that are critical to driving retinal growth and division. There are some empirical hints that this is the case. For example, it has been shown that RB1 loss makes retinal progenitor cells more dependent on estrogen-related receptor gamma (ESRRG) function. In turn, ESRRG then increases the expression of genes known to be critical retinal development: CRX, OTX2, NEUROD, and genes with LHX binding motifs. This may explain in part the retina-specific effects of RB1 disruption.
Conclusion
The study of retinoblastoma was instrumental in developing the modern paradigm of carcinogenesis, specifically substantiating Knudson’s 2-hit model. Unfortunately, the sporadic nature, especially the frequent contribution from de novo mutations, of retinoblastoma make it hard for genetic medicine to definitively resolve.10 However, early detection and treatment (surgical resection and sometimes chemotherapy) has been great for delivering high cure rates. Considering this, a future where universal whole-genome sequencing (WGS) is deployed may be one where no RB1 mutation carrier progresses to advanced disease. Plus, very early diagnosis may create a new window for early intervention. In fact, conjuring such scenarios in the consciousness of research scientists and clinicians may hasten its reality.
The incidence of additional malignancies is a concern among retinoblastoma patients but apart from reports of increased rates of skin cancer, there has yet to be a great definition of this presumably elevated risk.
There is a possibility that Pauw’s autopsy notes actually concerned an embryonal rhabdomyosarcoma instead of retinoblastoma. There is debate about this based on translations of the notes from Latin. However, Edwin B. Dunphy’s reading has set the narrative. Now, Pauw’s observations are forever connected with retinoblastoma regardless of the accuracy of the claim.
Roughly, 4.5% of retinoblastoma cases are somatic mosaicism according to a 2009 study.
We’ve grandfathered in plenty of misnomers though.
The actual structure of the RB1 gene was reported in PNAS by 1989. By this time however, this locus was widely understood to be the genetic instigation behind retinoblastoma.
The RB1 discovery work was also important for its secondary effects. It helped boost the scientific discovery effort on other tumor suppressor genes. It also helped shape how the field of cancer research thinks about how cancer develops.
RB1 carries out its repressive activity through a couple different mechanisms. This can include keeping E2F off regulatory regions of DNa but also by disrupting other transactivating factors from interacting with E2F while it is bound to regulatory DNA regions.
CDK4 is a cell cycle kinase that adds phosphate groups to RB1. The phosphorylation is the modification that removes RB1’s repression of E2F.
What I somewhat confusingly call organotropism to mean why certain gene mutations drive cancer in certain tissues appears to be a long-running mystery in cancer biology. At least, I’ve hoped to make this clear over the course of the Cancer Genomes series thus far.
Genetic engineering, gene therapy, or family planning would generally be not be deployable against RB1 de novo mutations, affecting only the 10-20% familial cases.